
Systematic Identification and Comparison of the Expressed Profiles of lncRNAs, miRNAs, circRNAs, and mRNAs with Associated Co-Expression Networks in Pigs with Low and High Intramuscular Fat


Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Animal and Sample Preparation
2.3. Construction and Sequencing of cDNA Libraries
2.4. Data Mapping and Transcriptome Assembly
2.5. Identification of lncRNAs, miRNAs, and circRNA
2.6. Differentially Expressed RNA Analysis
2.7. Prediction of the Potential Target Genes of DE lncRNAs, miRNAs, and circRNAs
2.8. Gene Ontology Enrichment and KEGG Pathway Analyses
2.9. Co-Construction of Gene Expression Networks
2.10. Association Analysis between QTL Sites and the Locations of Differentially Expressed RNA
2.11. Validation of the RNA Sequencing Results Using qRT-PCR
2.12. Statistical Analyses
3. Results
3.1. Overview of RNA Sequencing
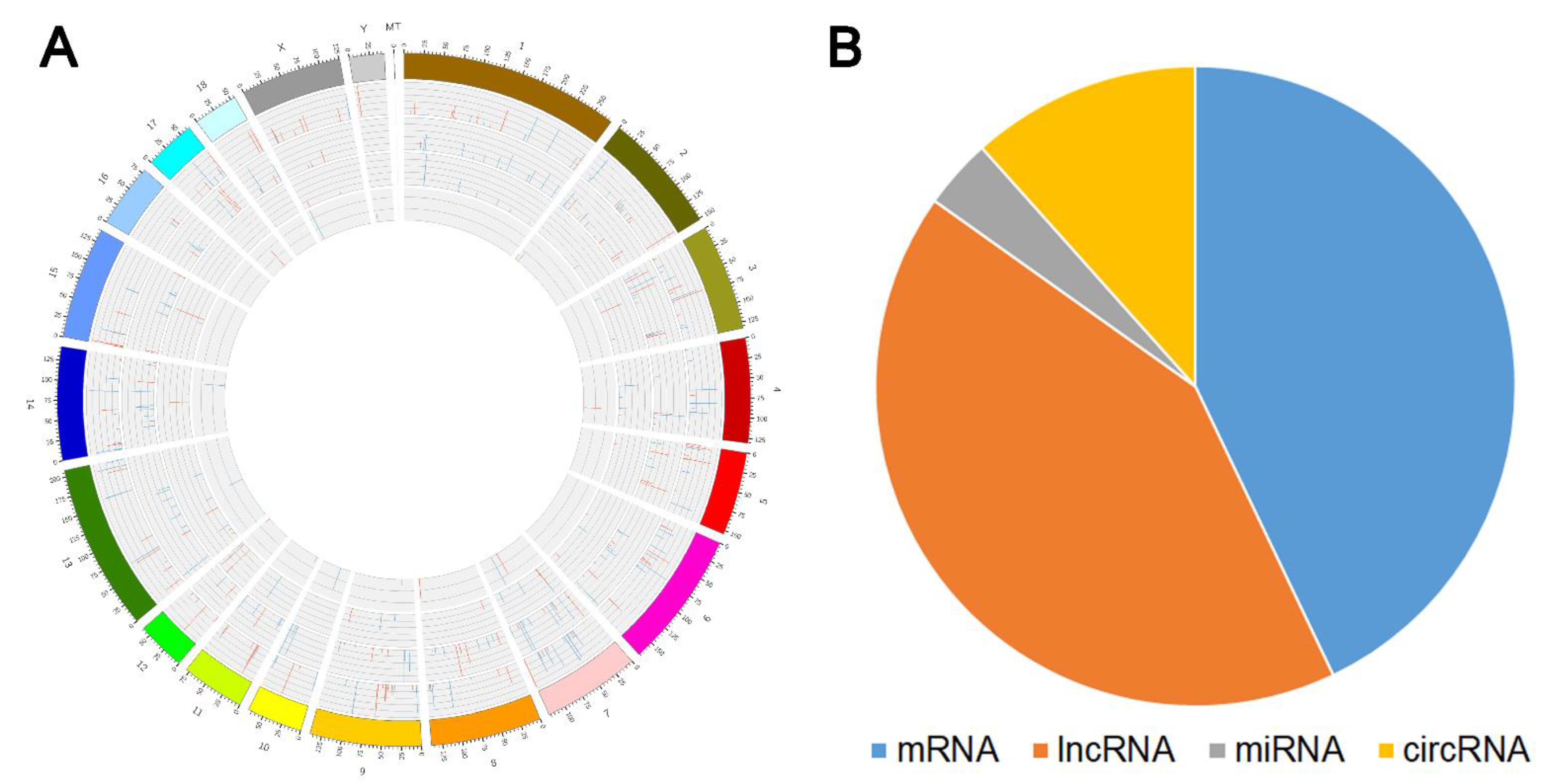
3.2. Differential Expression Profiles ofmRNAs lncRNAs, miRNAs, and circRNAs
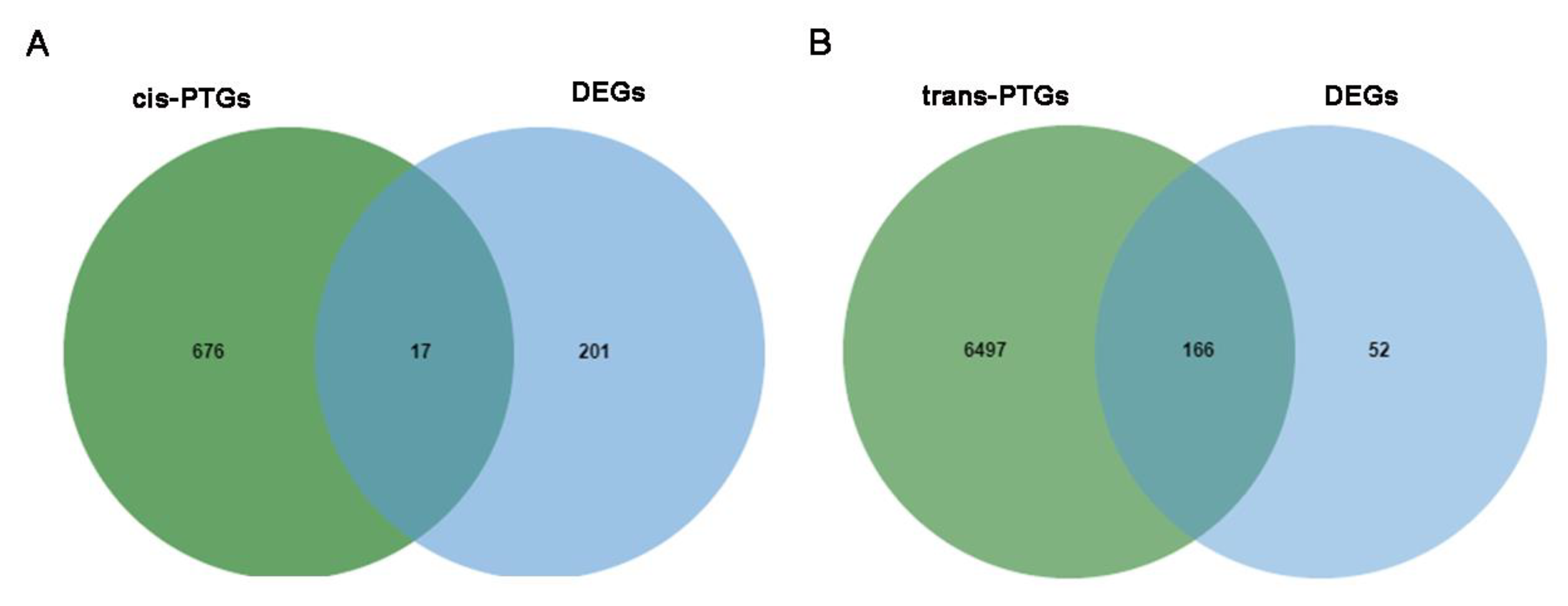
3.3. Prediction of the Potential Target Genes (PTGs) of DE lncRNAs, circRNAs, and miRNAs
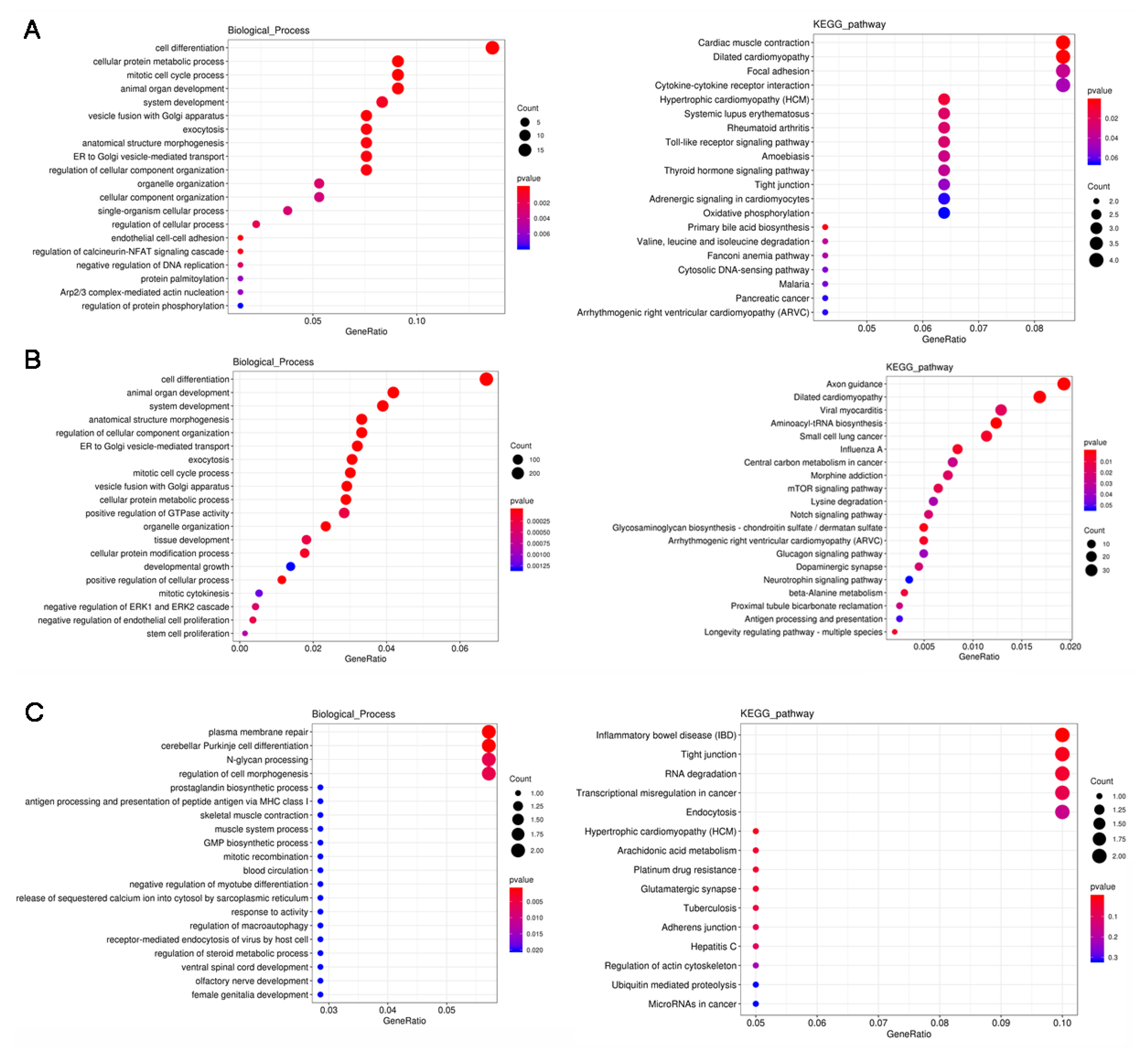
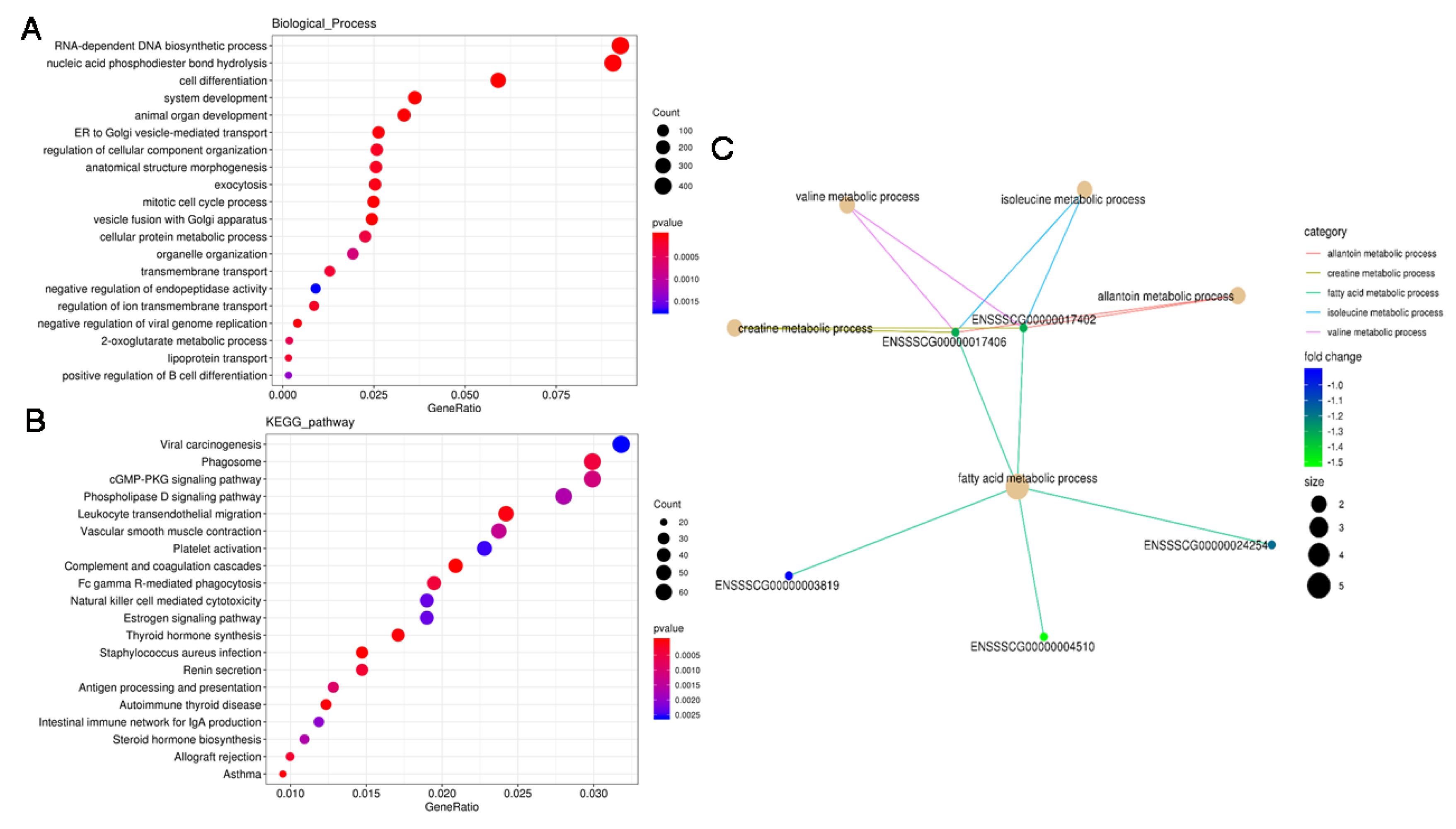
3.4. GO and KEGG Analysis of the DERs
3.5. Functional Analysis of the DE PTGs
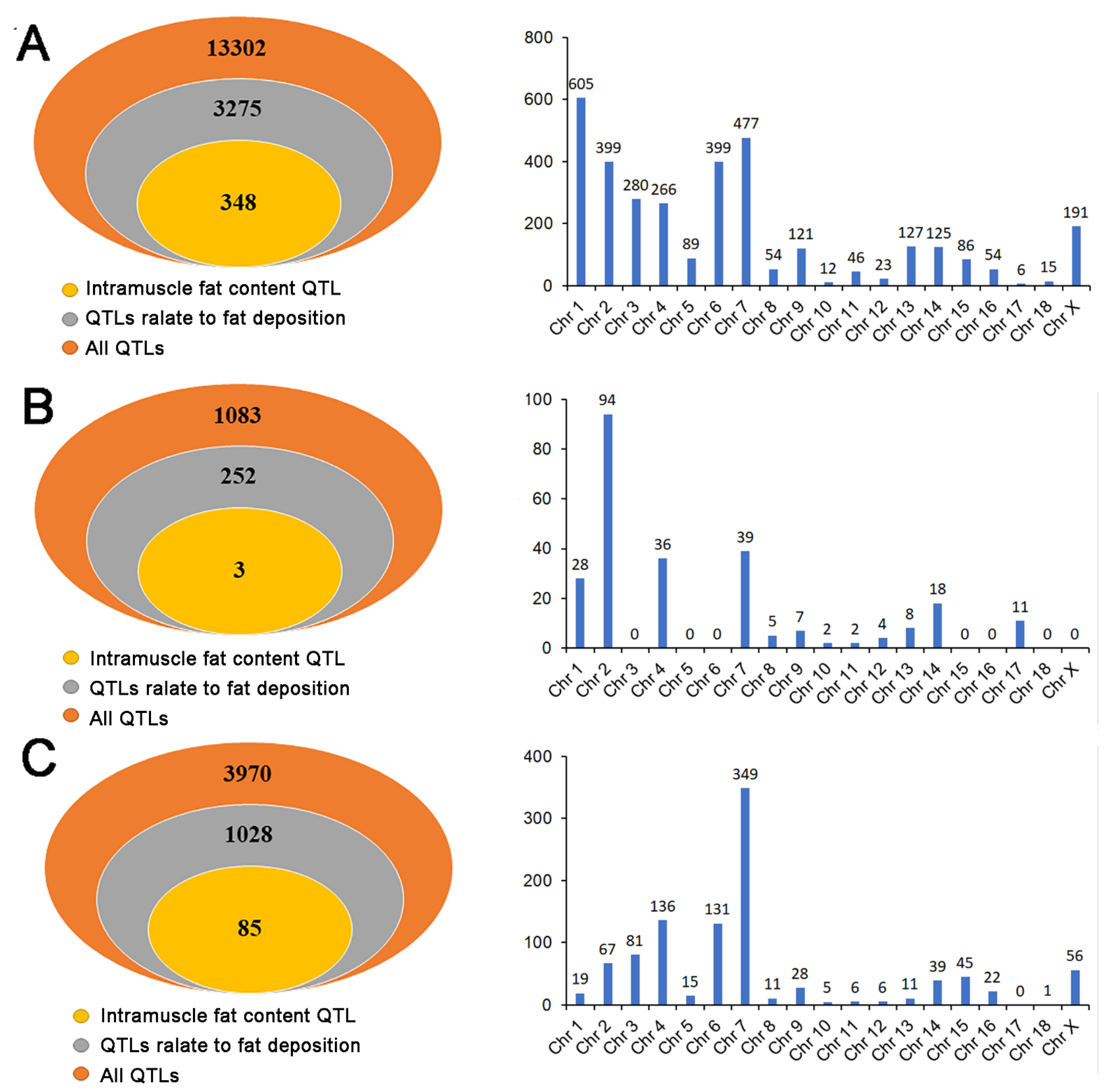
3.6. Overlapping Analysis between QTL Sites and the Location of DE RNAs
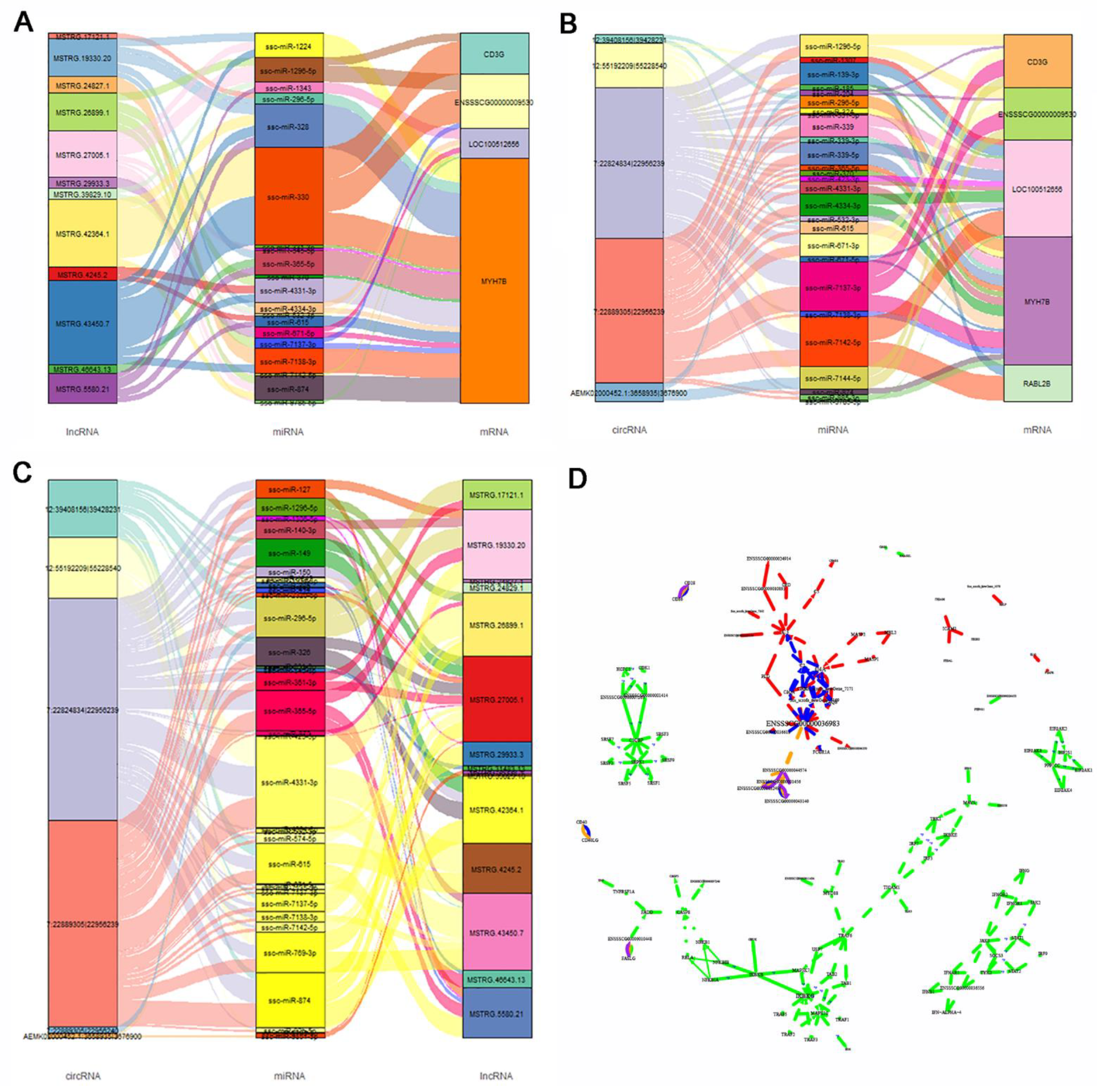
3.7. Expression Regulation Analysis of DE lncRNAs, miRNAs, and circRNAs, and Their DE PTGs
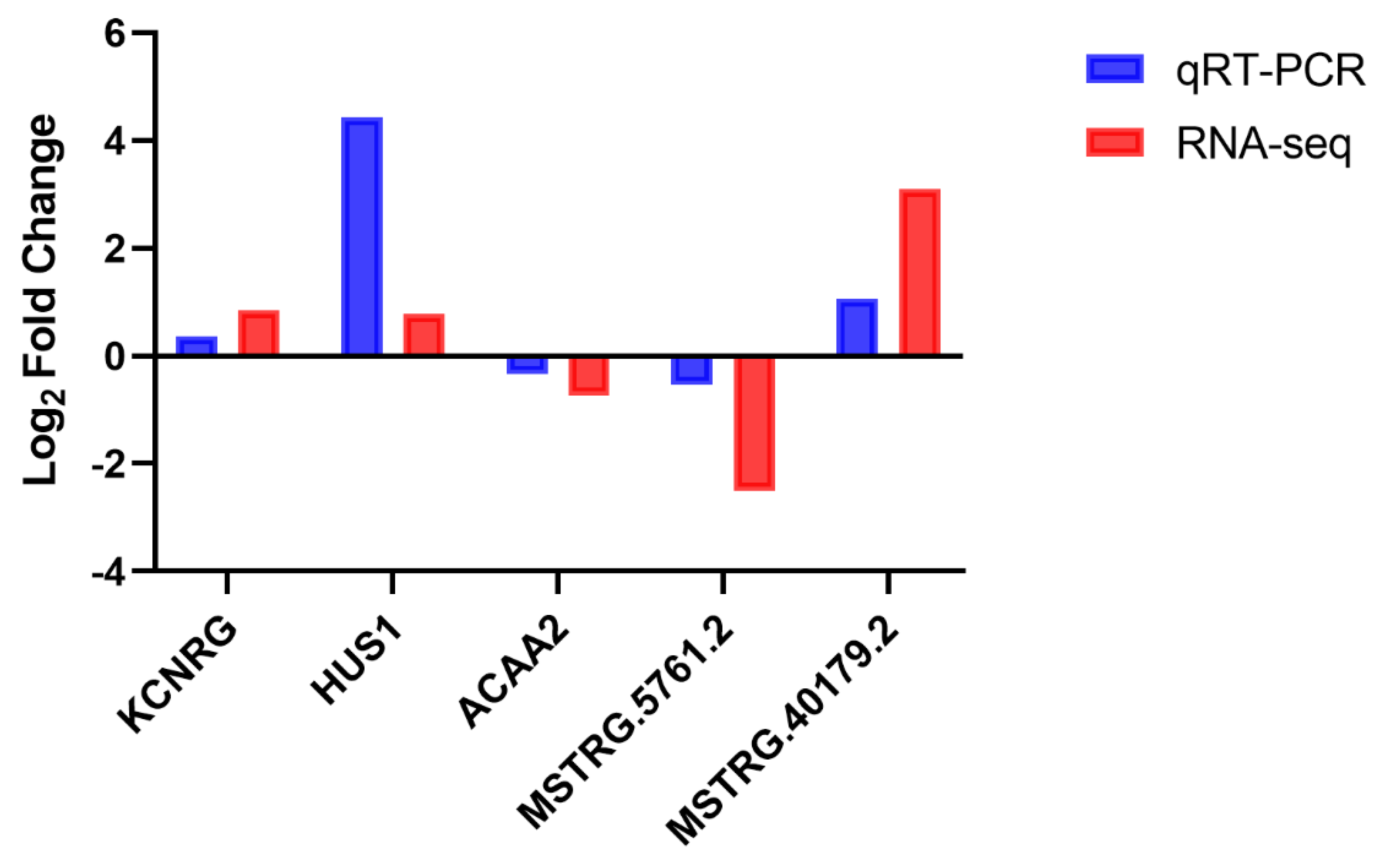
3.8. RNA Sequencing Results Validation Using qRT-PCR
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ΔΔCt | delta delta cycle threshold |
| ACAA2 | Acetyl-CoA Acyltransferase 2 |
| ACOX2 | Acyl-CoA oxidase 2 |
| CCL4 | C-C Motif Chemokine Ligand 4 |
| CCL10 | C-C Motif Chemokine Ligand 10 |
| CIB2 | calcium and integrin binding family member 2 |
| ceRNA | competing endogenous RNAs |
| circRNAs | circular RNAs |
| CXCL10 | C-X-C motif chemokine ligand 10 |
| CXCL16 | C-X-C motif chemokine ligand 16 |
| DE | Differential expression |
| DERs | differentially expressed RNAs |
| FPKM | Fragments per kilobase of exon model per million mapped fragments |
| GAPDH | Glyceraldehyde-3-phosphate dehydrogenase |
| GO | Gene ontology |
| GPAT | Glycerol-3-phosphate ethyltransferase |
| GPNMB | glycoprotein nmb. |
| GWAS | genome-wide association study |
| HUS1 | HUS1 Checkpoint Clamp Component |
| IMF | Intramuscular fat |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| KCNRG | Potassium Channel Regulator |
| lncRNAs | long non-coding RNAs |
| LDM | longissimus dorsi muscle |
| mRNAs | message RNA |
| miRNAs | microRNAs |
| miRNAs | microRNAs |
| MYH7B | myosin heavy chain 7B |
| Novel | novel gene |
| NPPC | National Pork Producers Council |
| PTGs | potential target genes |
| PTPMT1 | protein tyrosine phosphatase mitochondrial 1 |
| qRT-PCR | Reverse transcription quantitative polymerase chain reaction |
| QTLs | quantitative trait loci |
| RABL2B | RAB, member of ras oncogene family like 2b |
| RDH16 | retinol dehydrogenase 16 |
| rRNAs | mitochondrial ribosomal RNAs |
| SFRP4 | secreted frizzled-related protein 4 |
| sRNA | small RNA |
| snRNAs | small nuclear RNAs |
| snoRNA | small nucleolar RNA |
| SPP1 | secreted phosphoprotein 1 |
| TGFB3 | transforming growth factor beta 3 |
| THBS4 | thrombospondin 4 |
| tRNA | transfer RNA |
| MAPK10 | mitogen-activated protein kinase 10 |
| JAK1 | janus kinase 1 |
| STAT1 | signal transducer and activator of transcription 1 |
| TYK2 | tyrosine kinase 2 |
| IRF9 | interferon regulatory factor 9 |
| FADD | fas associated via death domain |
References
- Sans, P.; Combris, P. World meat consumption patterns: An overview of the last fifty years (1961–2011). Meat Sci. 2015, 109, 106–111. [Google Scholar] [CrossRef] [Green Version]
- Hwang, Y.-H.; Lee, S.-J.; Lee, E.-Y.; Joo, S.-T. Effects of carcass weight increase on meat quality and sensory properties of pork loin. J. Anim. Sci. Technol. 2020, 62, 753–760. [Google Scholar] [CrossRef]
- Brewer, M.; Zhu, L.; McKeith, F. Marbling effects on quality characteristics of pork loin chops: Consumer purchase intent, visual and sensory characteristics. Meat Sci. 2001, 59, 153–163. [Google Scholar] [CrossRef]
- Fortin, A.; Robertson, W.; Tong, A. The eating quality of Canadian pork and its relationship with intramuscular fat. Meat Sci. 2005, 69, 297–305. [Google Scholar] [CrossRef]
- Zhao, X.; Hu, H.; Lin, H.; Wang, C.; Wang, Y.; Wang, J. Muscle Transcriptome Analysis Reveals Potential Candidate Genes and Pathways Affecting Intramuscular Fat Content in Pigs. Front. Genet. 2020, 11, 877. [Google Scholar] [CrossRef]
- Grindflek, E.; Szyda, J.; Liu, Z.; Lien, S. Detection of quantitative trait loci for meat quality in a commercial slaughter pig cross. Mamm. Genome 2001, 12, 299–304. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Ren, J.; Guo, Y.; Duan, Y.; Ding, N.; Zhou, L.; Li, L.; Yan, X.; Yang, K.; Huang, L.; et al. Genome-wide identification of quantitative trait loci for carcass composition and meat quality in a large-scale White Duroc × Chinese Erhualian resource population. Anim. Genet. 2009, 40, 637–647. [Google Scholar] [CrossRef]
- Pearson, T.A. How to Interpret a Genome-wide Association Study. JAMA 2008, 299, 1335–1344. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Wang, J.; Yang, D.-D.; Liu, Z.-L.; Zeng, Y.-Q.; Chen, W. Expression of lipid metabolism genes provides new insights into intramuscular fat deposition in Laiwu pigs. Asian-Australas. J. Anim. Sci. 2020, 33, 390–397. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.X.; Zeng, Q.F.; Chen, W.; Zhang, H.; Zeng, Y.Q. Analysis and preliminary validation of the molecular mechanism of fat deposition in fatty and lean pigs by high-throughput sequencing. Mamm. Genome 2019, 30, 71–80. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Chen, K.; Wei, R.; Fan, G.; Cai, X.; Xu, L.; Cen, B.; Wang, J.; Xie, H.; Zheng, S.; et al. The circFASN/miR-33a pathway participates in tacrolimus-induced dysregulation of hepatic triglyceride homeostasis. Signal Transduct. Target. Ther. 2020, 5, 23. [Google Scholar] [CrossRef]
- Nawrocki, M.J.; Jopek, K.; Zdun, M.; Mozdziak, P.; Jemielity, M.; Perek, B.; Bukowska, D.; Kempisty, B. Expression Profile of Genes Encoding Proteins Involved in Regulation of Vasculature Development and Heart Muscle Morphogenesis—A Transcriptomic Approach Based on a Porcine Model. Int. J. Mol. Sci. 2021, 22, 8794. [Google Scholar] [CrossRef]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef]
- Pertea, M.; Kim, D.; Pertea, G.M.; Leek, J.T.; Salzberg, S.L. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 2016, 11, 1650–1667. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kong, L.; Zhang, Y.E.; Ye, Z.-Q.; Liu, X.-Q.; Zhao, S.-Q.; Wei, L.; Gao, G. CPC: Assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Res. 2007, 35, W345–W349. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Luo, H.; Bu, D.; Zhao, G.; Yu, K.; Zhang, C.; Liu, Y.; Chen, R.; Zhao, Y. Utilizing sequence intrinsic composition to classify protein-coding and long non-coding transcripts. Nucleic Acids Res. 2013, 41, e166. [Google Scholar] [CrossRef]
- Wang, L.; Park, H.J.; Dasari, S.; Wang, S.; Kocher, J.-P.; Li, W. CPAT: Coding-Potential Assessment Tool using an alignment-free logistic regression model. Nucleic Acids Res. 2013, 41, e74. [Google Scholar] [CrossRef] [PubMed]
- Friedlander, M.; Mackowiak, S.; Li, N.; Chen, W.; Rajewsky, N. miRDeep2 accurately identifies known and hundreds of novel microRNA genes in seven animal clades. Nucleic Acids Res. 2011, 40, 37–52. [Google Scholar] [CrossRef]
- Gao, Y.; Wang, J.; Zhao, F. CIRI: An efficient and unbiased algorithm for de novo circular RNA identification. Genome Biol. 2015, 16, 1–16. [Google Scholar] [CrossRef] [Green Version]
- I Love, M.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, M.D.; McCarthy, D.; Smyth, G. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2009, 26, 139–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Betel, D.; Wilson, M.; Gabow, A.; Marks, D.S.; Sander, C. The microRNA.org resource: Targets and expression. Nucleic Acids Res. 2007, 36, D149–D153. [Google Scholar] [CrossRef] [Green Version]
- Lewis, B.P.; Shih, I.-H.; Jones-Rhoades, M.W.; Bartel, D.P.; Burge, C.B. Prediction of Mammalian MicroRNA Targets. Cell 2003, 115, 787–798. [Google Scholar] [CrossRef] [Green Version]
- Yu, G.; Wang, L.-G.; Han, Y.; He, Q.-Y. clusterProfiler: An R Package for Comparing Biological Themes Among Gene Clusters. OMICS A J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Gan, M.; Tang, T.; Shao, J.; Lai, T.; Ma, Y.; Elzo, M.A.; Jia, X.; Hu, S.; Wang, J.; et al. Intramuscular adipocyte and fatty acid differences between high-fat and control rabbit groups subject to a restricted diet. Veter. Med. Sci. 2021, 7, 2051–2060. [Google Scholar] [CrossRef]
- Hocquette, J.F.; Gondret, F.; Baéza, E.; Médale, F.; Jurie, C.; Pethick, D.W. Intramuscular fat content in meat-producing animals: Development, genetic and nutritional control, and identification of putative markers. Animal 2010, 4, 303–319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blanchard, P.J.; Willis, M.B.; Warkup, C.C.; Ellis, M. The influence of carcass backfat and intramuscular fat level on pork eating quality. J. Sci. Food Agric. 2000, 80, 145–151. [Google Scholar] [CrossRef]
- Wang, Y.; Ma, C.; Sun, Y.; Li, Y.; Kang, L.; Jiang, Y. Dynamic transcriptome and DNA methylome analyses on longissimus dorsi to identify genes underlying intramuscular fat content in pigs. BMC Genom. 2017, 18, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Zhong, J.; Zhang, C.; Chai, Z.; Cao, H.; Wang, J.; Zhu, J.; Wang, J.; Ji, Q. The whole-transcriptome landscape of muscle and adipose tissues reveals the ceRNA regulation network related to intramuscular fat deposition in yak. BMC Genom. 2020, 21, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Zappaterra, M.; Gioiosa, S.; Chillemi, G.; Zambonelli, P.; Davoli, R. Muscle transcriptome analysis identifies genes involved in ciliogenesis and the molecular cascade associated with intramuscular fat content in Large White heavy pigs. PLoS ONE 2020, 15, e0233372. [Google Scholar] [CrossRef]
- Yuan, Z.; Li, J.; Li, J.; Gao, X.; Gao, H.; Xu, S. Effects of DGAT1 gene on meat and carcass fatness quality in Chinese commercial cattle. Mol. Biol. Rep. 2012, 40, 1947–1954. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Zhou, Y.; Nong, Q.; You, W.; Wang, L.; Wang, Y.; Shan, T. LKB1 Differently Regulates Adipogenesis in Intramuscular and Subcutaneous Adipocytes through Metabolic and Cytokine-Related Signaling Pathways. Cells 2020, 9, 2599. [Google Scholar] [CrossRef]
- Du, Y.; Wang, Y.; Xu, Q.; Zhu, J.; Lin, Y. TMT-based quantitative proteomics analysis reveals the key proteins related with the differentiation process of goat intramuscular adipocytes. BMC Genom. 2021, 22, 1–14. [Google Scholar] [CrossRef]
- Jeong, J.Y.; Kim, J.S.; Nguyen, T.H.; Lee, H.-J.; Baik, M. Wnt/β-catenin signaling and adipogenic genes are associated with intramuscular fat content in the longissimus dorsi muscle of Korean cattle. Anim. Genet. 2013, 44, 627–635. [Google Scholar] [CrossRef]
- Hörbelt, T.; Knebel, B.; Fahlbusch, P.; Barbosa, D.; de Wiza, D.H.; Van de Velde, F.; Van Nieuwenhove, Y.; Lapauw, B.; Thoresen, G.H.; Al-Hasani, H.; et al. The adipokine sFRP4 induces insulin resistance and lipogenesis in the liver. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2019, 1865, 2671–2684. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, K.; Reue, K. Biochemistry, physiology, and genetics of GPAT, AGPAT, and lipin enzymes in triglyceride synthesis. Am. J. Physiol.-Endocrinol. Metab. 2009, 296, E1195–E1209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shan, D.; Li, J.L.; Wu, L.; Li, D.; Hurov, J.; Tobin, J.F.; Gimeno, R.E.; Cao, J. GPAT3 and GPAT4 are regulated by insulin-stimulated phosphorylation and play distinct roles in adipogenesis [S]. J. Lipid Res. 2010, 51, 1971–1981. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Fang, X.; Yang, R.; Yu, H.; Jiang, P.; Sun, B.; Zhao, Z. MiR-152 Regulates Apoptosis and Triglyceride Production in MECs via Targeting ACAA2 and HSD17B12 Genes. Sci. Rep. 2018, 8, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Petrus, P.; Mejhert, N.; Corrales, P.; Lecoutre, S.; Li, Q.; Maldonado, E.; Kulyté, A.; Lopez, Y.; Campbell, M.; Acosta, J.R.; et al. Transforming Growth Factor-β3 Regulates Adipocyte Number in Subcutaneous White Adipose Tissue. Cell Rep. 2018, 25, 551–560.e5. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Cui, H.; Fu, R.; Zheng, M.; Liu, R.; Zhao, G.; Wen, J. The regulation of IMF deposition in pectoralis major of fast- and slow- growing chickens at hatching. J. Anim. Sci. Biotechnol. 2017, 8, 77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Q.; Peng, J.; Chen, X.; Li, H.; Song, M.; Cheng, B.; Wu, T. Obesity and genes related to lipid metabolism predict poor survival in oral squamous cell carcinoma. Oral Oncol. 2018, 89, 14–22. [Google Scholar] [CrossRef]
- Lv, L.; Liang, W.; Ye, M.; Zhang, J.; Zhang, H.; Xue, G.; Zhang, L. Thrombospondin-4 ablation reduces macrophage recruitment in adipose tissue and neointima and suppresses injury-induced restenosis in mice. Atherosclerosis 2016, 247, 70–77. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Tai, L.; Zhang, L.; Chu, Y.; Li, Y.; Zhou, L. Comparative analyses of long non-coding RNA in lean and obese pigs. Oncotarget 2017, 8, 41440–41450. [Google Scholar] [CrossRef] [PubMed]
- Miao, Z.; Wang, S.; Zhang, J.; Wei, P.; Guo, L.; Liu, D.; Wang, Y.; Shi, M. Identification and comparison of long non-conding RNA in Jinhua and Landrace pigs. Biochem. Biophys. Res. Commun. 2018, 506, 765–771. [Google Scholar] [CrossRef]
- Xing, K.; Wang, K.; Ao, H.; Chen, S.; Tan, Z.; Wang, Y.; Xitong, Z.; Yang, T.; Zhang, F.; Liu, Y.; et al. Comparative adipose transcriptome analysis digs out genes related to fat deposition in two pig breeds. Sci. Rep. 2019, 9, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Tang, Z.; Wu, Y.; Yang, Y.; Yang, Y.-C.T.; Wang, Z.; Yuan, J.; Yang, Y.; Hua, C.; Fan, X.; Niu, G.; et al. Comprehensive analysis of long non-coding RNAs highlights their spatio-temporal expression patterns and evolutional conservation in Sus scrofa. Sci. Rep. 2017, 7, srep43166. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Huang, Z.; Zhao, W.; Li, M.; Li, C. Transcriptome Analysis Reveals Long Intergenic Non-Coding RNAs Contributed to Intramuscular Fat Content Differences between Yorkshire and Wei Pigs. Int. J. Mol. Sci. 2020, 21, 1732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zou, C.; Li, S.; Deng, L.; Guan, Y.; Chen, D.; Yuan, X.; Xia, T.; He, X.; Shan, Y.; Li, C. Transcriptome Analysis Reveals Long Intergenic Noncoding RNAs Contributed to Growth and Meat Quality Differences between Yorkshire and Wannanhua Pig. Genes 2017, 8, 203. [Google Scholar] [CrossRef] [Green Version]
- Zeng, Y.; Wang, G.; Zhou, C.-F.; Zhang, H.-B.; Sun, H.; Zhang, W.; Zhou, H.; Liu, R.; Zhu, Y.-S. LncRNA Profile Study Reveals a Three-LncRNA Signature Associated with the Pathological Complete Response Following Neoadjuvant Chemotherapy in Breast Cancer. Front. Pharmacol. 2019, 10, 574. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Tolleson, W.H.; Yu, D.; Chen, S.; Guo, L.; Xiao, W.; Tong, W.; Ning, B. Regulation of cytochrome P450 expression by microRNAs and long noncoding RNAs: Epigenetic mechanisms in environmental toxicology and carcinogenesis. J. Environ. Sci. Heal. Part C 2019, 37, 180–214. [Google Scholar] [CrossRef]
- Di Ciaula, A.; Garruti, G.; Baccetto, R.; Molina, E.M.; Bonfrate, L.; Wang, D.Q.-H.; Portincasa, P. Bile Acid Physiology. Ann. Hepatol. 2017, 16, S4–S14. [Google Scholar] [CrossRef]
- Gan, Z.; Rumsey, J.; Hazen, B.C.; Lai, L.; Leone, T.C.; Vega, R.B.; Xie, H.; Conley, K.E.; Auwerx, J.; Smith, S.R.; et al. Nuclear receptor/microRNA circuitry links muscle fiber type to energy metabolism. J. Clin. Investig. 2013, 123, 2564–2575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iqbal, A.; Ping, J.; Ali, S.; Zhen, G.; Juan, L.; Kang, J.Z.; Yi, P.Z.; Huixian, L.; Zhihui, Z. Role of microRNAs in myogenesis and their effects on meat quality in pig —A review. Asian-Australas. J. Anim. Sci. 2020, 33, 1873–1884. [Google Scholar] [CrossRef]
- Xu, M.; Zheng, X.; Jiang, F.; Qiu, W. MicroRNA-190b regulates lipid metabolism and insulin sensitivity by targeting IGF-1 and ADAMTS9 in non-alcoholic fatty liver disease. J. Cell. Biochem. 2018, 119, 5864–5874. [Google Scholar] [CrossRef]
- Chen, F.-F.; Xiong, Y.; Peng, Y.; Gao, Y.; Qin, J.; Chu, G.-Y.; Pang, W.-J.; Yang, G.-S. miR-425-5p Inhibits Differentiation and Proliferation in Porcine Intramuscular Preadipocytes. Int. J. Mol. Sci. 2017, 18, 2101. [Google Scholar] [CrossRef] [Green Version]
- Du, J.; Xu, Y.; Zhang, P.; Zhao, X.; Gan, M.; Li, Q.; Ma, J.; Tang, G.; Jiang, Y.; Wang, J.; et al. MicroRNA-125a-5p Affects Adipocytes Proliferation, Differentiation and Fatty Acid Composition of Porcine Intramuscular Fat. Int. J. Mol. Sci. 2018, 19, 501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.; Wang, T.; Chen, X.; Jiang, J.; Song, N.; Li, R.; Xin, Y.; Xuan, S. Inhibition of miR-499-5p expression improves nonalcoholic fatty liver disease. Ann. Hum. Genet. 2020, 1–10. [Google Scholar] [CrossRef]
- Sun, N.; Shen, C.; Zhang, L.; Wu, X.; Yu, Y.; Yang, X.; Yang, C.; Zhong, C.; Gao, Z.; Miao, W.; et al. Hepatic Krüppel-like factor 16 (KLF16) targets PPARα to improve steatohepatitis and insulin resistance. Gut 2020, 70, 2183–2195. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.-H.; Ho, K.-T.; Lu, S.-H.; Huang, C.-N.; Shiau, M.-Y. Regulation of glucose/lipid metabolism and insulin sensitivity by interleukin-4. Int. J. Obes. 2011, 36, 993–998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dutra, F.; Teixeira, L.; Costa, M.F.D.S.; Bozza, P.T. Fat, fight, and beyond: The multiple roles of lipid droplets in infections and inflammation. J. Leukoc. Biol. 2019, 106, 563–580. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Fahrmann, J.F.; Lee, H.; Li, Y.-J.; Tripathi, S.; Yue, C.; Zhang, C.; Lifshitz, V.; Song, J.; Yuan, Y.; et al. JAK/STAT3-Regulated Fatty Acid β-Oxidation Is Critical for Breast Cancer Stem Cell Self-Renewal and Chemoresistance. Cell Metab. 2017, 27, 136–150.e5. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.Z.; Zhang, Z.M.; Zhou, L.T.; Zhu, J.; Zhang, X.H.; Qi, W.; Ding, S.; Xu, Q.; Han, X.; Zhao, Y.M.; et al. Di (2-ethylhexyl) phthalate Disorders Lipid Metabolism via TYK2/STAT1 and Autophagy in Rats. Environ. Sci. 2019, 32, 406–418. [Google Scholar]
- Grunert, T.; Leitner, N.R.; Marchetti-Deschmann, M.; Miller, I.; Wallner, B.; Radwan, M.; Vogl, C.; Kolbe, T.; Kratky, D.; Gemeiner, M.; et al. A comparative proteome analysis links tyrosine kinase 2 (Tyk2) to the regulation of cellular glucose and lipid metabolism in response to poly(I:C). J. Proteom. 2011, 74, 2866–2880. [Google Scholar] [CrossRef] [Green Version]
- Zhuang, H.; Wang, X.; Zha, D.; Gan, Z.; Cai, F.; Du, P.; Yang, Y.; Yang, B.; Zhang, X.; Yao, C.; et al. FADD is a key regulator of lipid metabolism. EMBO Mol. Med. 2016, 8, 895–918. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Carcass Weight (kg) | IMF (%) | Group |
|---|---|---|---|
| H1 | 82.6 | 4.07 | High IMF |
| H2 | 113 | 4.40 | High IMF |
| H3 | 113 | 4.56 | High IMF |
| H4 | 97.4 | 4.98 | High IMF |
| H5 | 96.8 | 5.43 | High IMF |
| L1 | 92 | 1.05 | Low IMF |
| L2 | 113 | 1.18 | Low IMF |
| L3 | 125.6 | 1.28 | Low IMF |
| L4 | 89.4 | 1.57 | Low IMF |
| L5 | 94.4 | 1.60 | Low IMF |
| RNA | Regulated | log2FC | p-Value | Type |
|---|---|---|---|---|
| MSTRG.19330.20 | up | 6.573623 | 5.41 × 10−62 | lncRNAs |
| MSTRG.40179.2 | up | 3.10731 | 1.32 × 10−10 | |
| MSTRG.44176.8 | up | 1.908359 | 2.75 × 10−5 | |
| MSTRG.39829.10 | up | 1.710734 | 4.15 × 10−4 | |
| MSTRG.38601.10 | up | 1.476951 | 1.54 × 10−6 | |
| MSTRG.29140.1 | down | −1.94114 | 2.37 × 10−5 | |
| MSTRG.25219.1 | down | −1.99459 | 2.95 × 10−5 | |
| MSTRG.9199.1 | down | −2.01282 | 4.72 × 10−6 | |
| MSTRG.5761.2 | down | −2.50226 | 3.96 × 10−8 | |
| MSTRG.44725.16 | down | −2.71452 | 1.45 × 10−10 | |
| novel_miR_118 | up | 1.459799 | 1.07 × 10−2 | miRNAs |
| ssc-miR-208b | up | 1.310524 | 7.14 × 10−3 | |
| novel_miR_398 | up | 1.297012 | 3.44 × 10−2 | |
| novel_miR_278 | up | 1.272563 | 3.34 × 10−2 | |
| ssc-miR-190b | up | 1.198435 | 1.12 × 10−2 | |
| ssc-miR-499-5p | up | 1.185263 | 1.01 × 10−2 | |
| novel_miR_185 | down | −1.54969 | 1.09 × 10−2 | |
| novel_miR_45 | down | −1.74161 | 3.76 × 10−3 | |
| novel_miR_476 | down | −1.78496 | 3.59 × 10−3 | |
| novel_miR_45 | down | −1.74161 | 3.76 × 10−3 | |
| novel_miR_476 | down | −1.78496 | 3.59 × 10−3 | |
| 12:39408156|39428231 | up | 9.005799 | 1.98 × 10−6 | circRNAs |
| 14:71348983|71349948 | up | 6.965969 | 4.78 × 10−4 | |
| 3:44121881|44122061 | up | 6.819521 | 3.29 × 10−4 | |
| 9:125732918|125735258 | up | 6.586107 | 1.03 × 10−3 | |
| 13:71794794|71797638 | up | 6.346968 | 1.25 × 10−3 | |
| 1:108385212|108386218 | down | −5.96632 | 1.61 × 10−3 | |
| 12:59320434|59323398 | down | −6.16306 | 1.81 × 10−3 | |
| 7:68514625|68532510 | down | −6.37264 | 1.20 × 10−3 | |
| 9:66405629|66409132 | down | −6.57113 | 8.32 × 10−4 | |
| 4:50433434|50447885 | down | −6.83942 | 4.52 × 10−4 | |
| RDH16 | up | 0.993121 | 1.20 × 10−4 | mRNAs |
| ENSSSCG00000045560 | up | 0.928328 | 2.29 × 10−4 | |
| KCNRG | up | 0.856391 | 6.58 × 10−4 | |
| ENSSSCG00000045892 | up | 0.844323 | 8.67 × 10−4 | |
| RABL2B | up | 0.831953 | 1.16 × 10−3 | |
| SPP1 | down | −0.92796 | 2.69 × 10−4 | |
| CIB2 | down | −0.93599 | 8.18 × 10−5 | |
| PTPMT1 | down | −0.96233 | 5.00 × 10−5 | |
| MYH7B | down | −0.99074 | 1.05 × 10−5 | |
| GPNMB | down | −1.15542 | 1.07 × 10−5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cheng, F.; Liang, J.; Yang, L.; Lan, G.; Wang, L.; Wang, L. Systematic Identification and Comparison of the Expressed Profiles of lncRNAs, miRNAs, circRNAs, and mRNAs with Associated Co-Expression Networks in Pigs with Low and High Intramuscular Fat. Animals 2021, 11, 3212. https://doi.org/10.3390/ani11113212
Cheng F, Liang J, Yang L, Lan G, Wang L, Wang L. Systematic Identification and Comparison of the Expressed Profiles of lncRNAs, miRNAs, circRNAs, and mRNAs with Associated Co-Expression Networks in Pigs with Low and High Intramuscular Fat. Animals. 2021; 11(11):3212. https://doi.org/10.3390/ani11113212
Chicago/Turabian StyleCheng, Feng, Jing Liang, Liyu Yang, Ganqiu Lan, Lixian Wang, and Ligang Wang. 2021. "Systematic Identification and Comparison of the Expressed Profiles of lncRNAs, miRNAs, circRNAs, and mRNAs with Associated Co-Expression Networks in Pigs with Low and High Intramuscular Fat" Animals 11, no. 11: 3212. https://doi.org/10.3390/ani11113212