
Genomic Diversity of CRESS DNA Viruses in the Eukaryotic Virome of Swine Feces


 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Processing and Random PCR for Metagenomics
2.2. Sequencing
2.3. Amplification Of Complete Viral Sequences
2.4. Software
3. Results
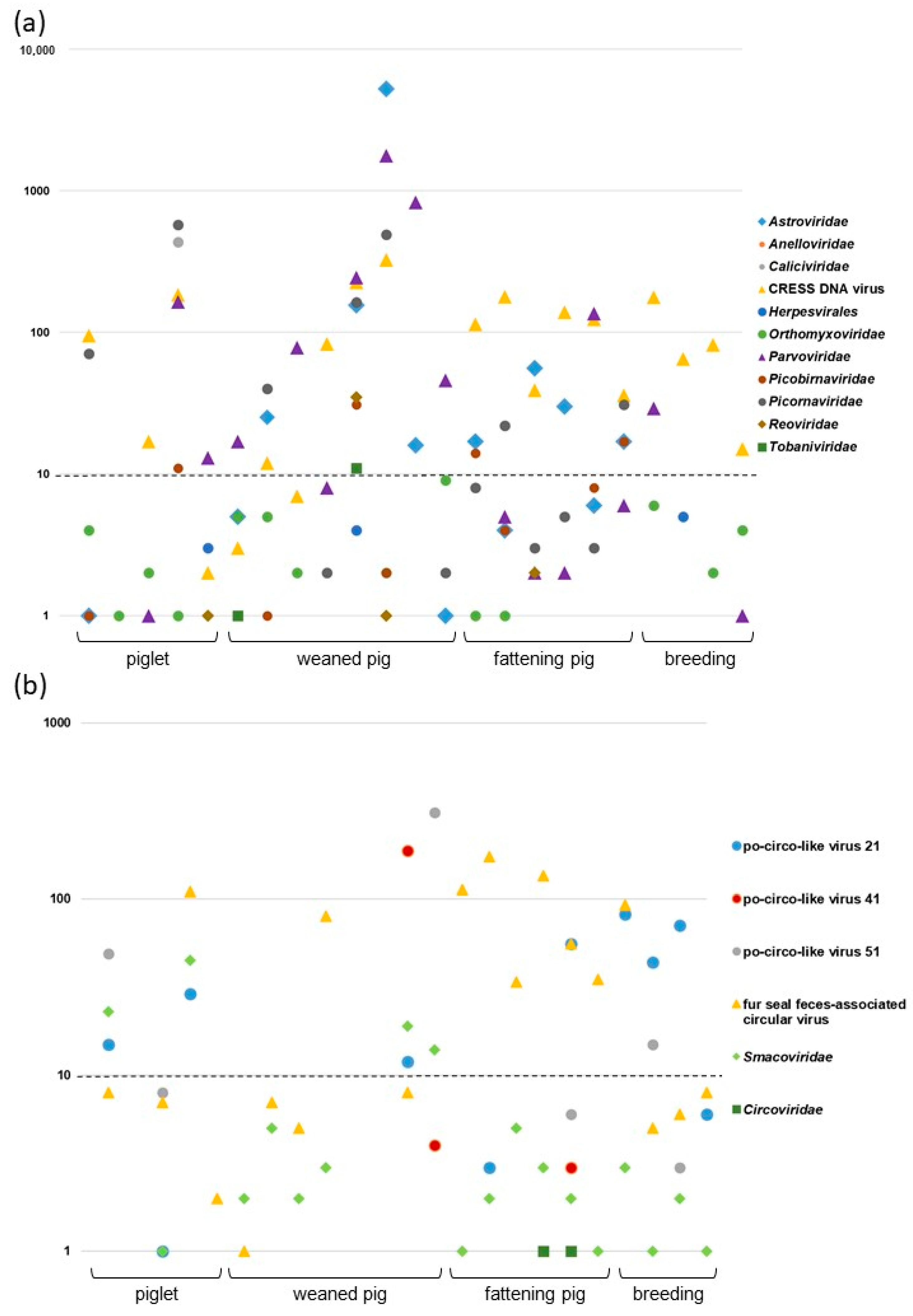
3.1. Results of Viral Metagenomics and Focus on CRESS DNA Viruses
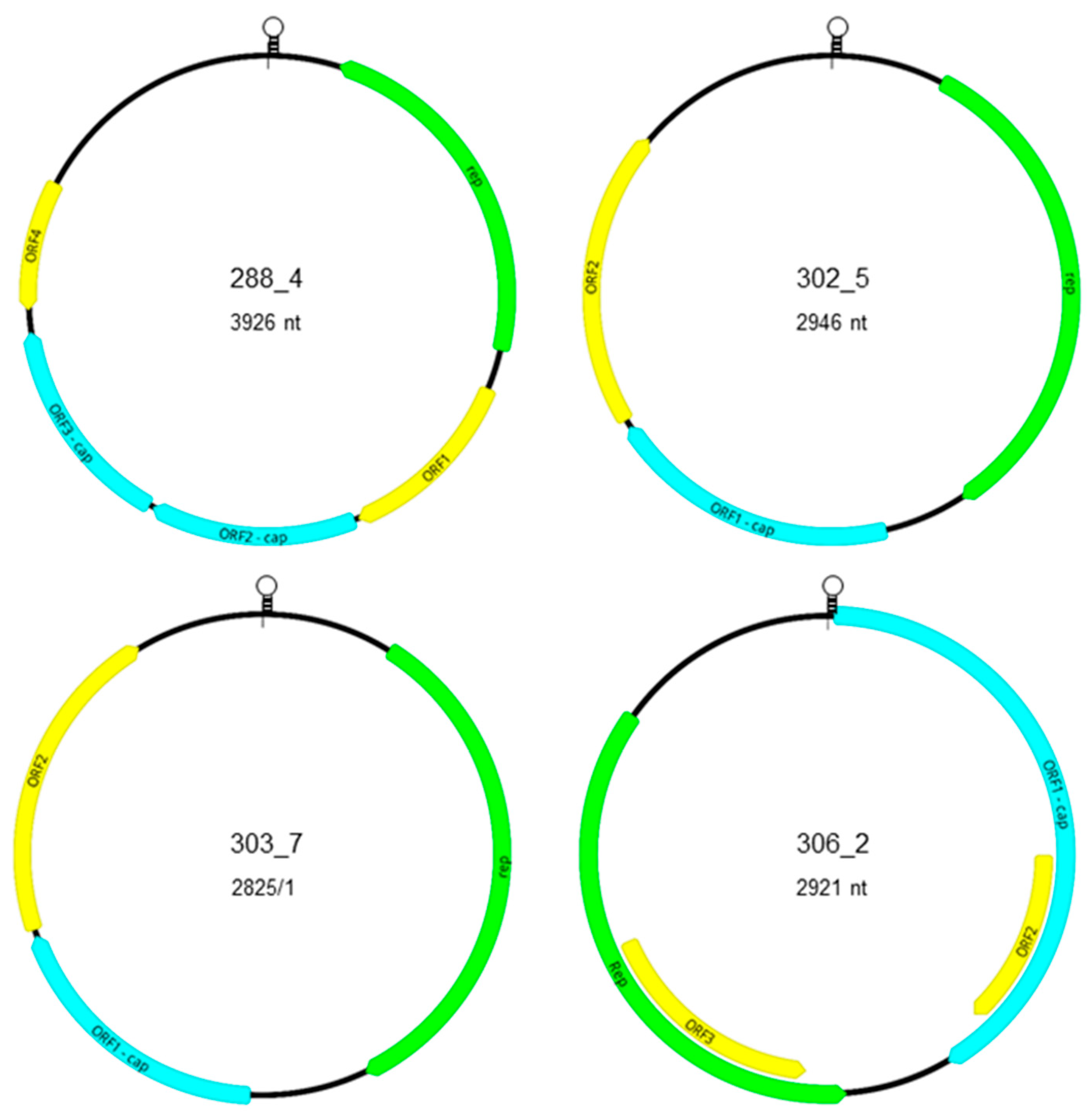
3.2. Genomic Characterization of Novel Po-Circo-Like CRESS DNA Viruses
3.3. Genomic Characterization of Novel FSfaCV-Like CRESS DNA Viruses
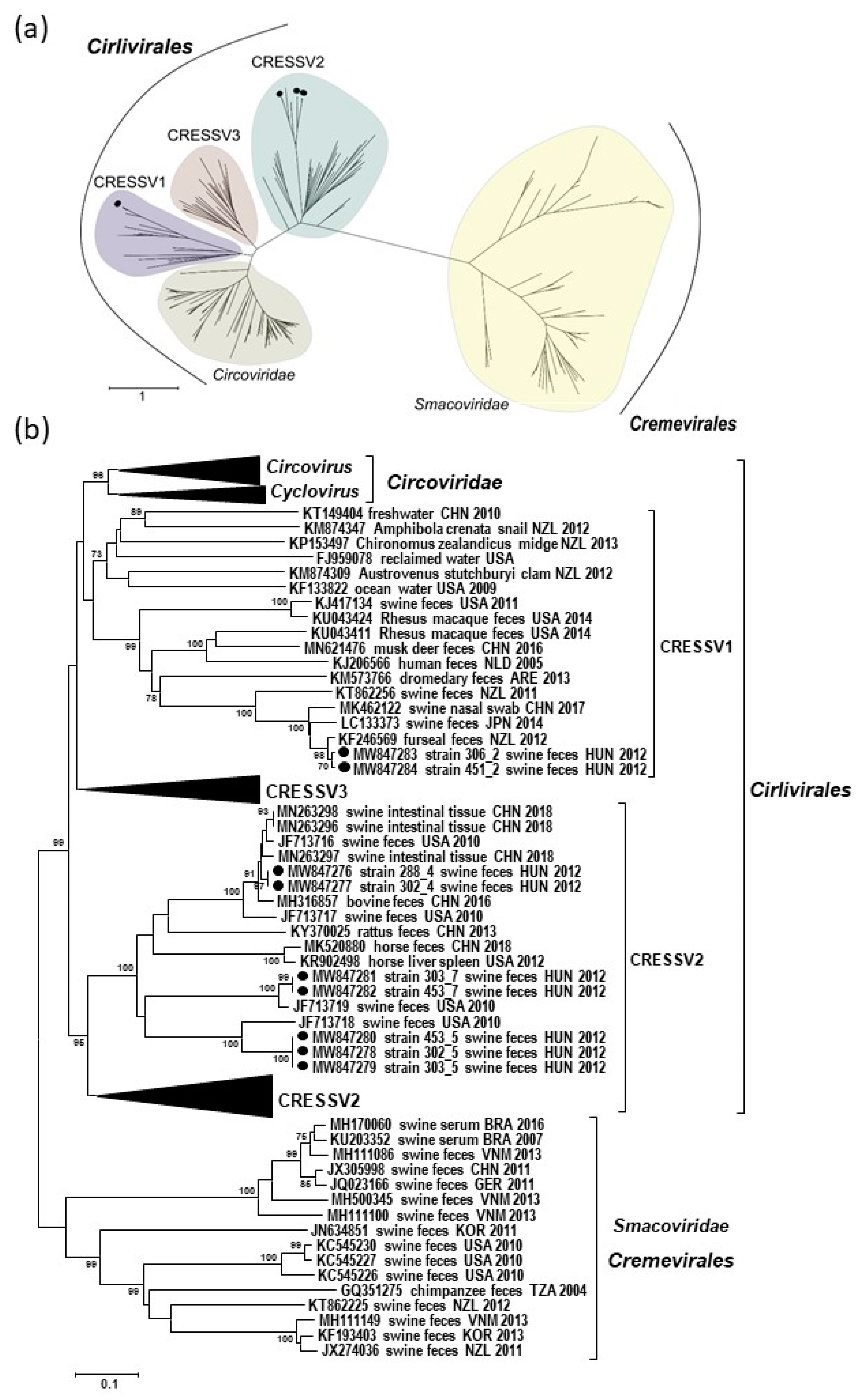
3.4. Phylogenetic Classification of the Genomes of Novel Porcine CRESS DNA Viruses
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kazlauskas, D.; Varsani, A.; Koonin, E.V.; Krupovic, M. Multiple origins of prokaryotic and eukaryotic single-stranded DNA viruses from bacterial and archaeal plasmids. Nat. Commun. 2019, 10, 3425. [Google Scholar] [CrossRef] [Green Version]
- Zhao, L.; Rosario, K.; Breitbart, M.; Duffy, S. Eukaryotic circular Rep-encoding single-stranded DNA (CRESS DNA) viruses: Ubiquitous viruses with small genomes and a diverse host range. Adv. Virus. Res. 2019, 103, 71–133. [Google Scholar] [CrossRef]
- Krupovic, M.; Varsani, A.; Kazlauskas, D.; Breitbart, M.; Delwart, E.; Rosario, K.; Yutin, N.; Wolf, Y.I.; Harrach, B.; Zerbini, F.M.; et al. Cressdnaviricota: A virus phylum unifying seven families of Rep-encoding viruses with single-stranded, circular DNA genomes. J. Virol. 2020, 94, e00582-20. [Google Scholar] [CrossRef]
- Rosario, K.; Duffy, S.; Breitbart, M. A field guide to eukaryotic circular single-stranded DNA viruses: Insights gained from metagenomics. Arch. Virol. 2012, 157, 1851–1871. [Google Scholar] [CrossRef] [PubMed]
- Fehér, E.; Székely, C.; Lőrincz, M.; Cech, G.; Tuboly, T.; Singh, H.S.; Bányai, K.; Farkas, S.L. Integrated circoviral rep-like sequences in the genome of cyprinid fish. Virus Genes 2013, 47, 374–377. [Google Scholar] [CrossRef] [PubMed]
- Marton, S.; Ihász, K.; Lengyel, G.; Farkas, S.L.; Dán, Á.; Paulus, P.; Bányai, K.; Fehér, E. Ubiquiter circovirus sequences raise challenges in laboratory diagnosis: The case of honey bee and bee mite, reptiles, and free living amoebae. Acta Microbiol. Immunol. Hung. 2015, 62, 57–73. [Google Scholar] [CrossRef] [Green Version]
- Rosario, K.; Breitbart, M.; Harrach, B.; Segalés, J.; Delwart, E.; Biagini, P.; Varsani, A. Revisiting the taxonomy of the family Circoviridae: Establishment of the genus Cyclovirus and removal of the genus Gyrovirus. Arch. Virol. 2017, 162, 1447–1463. [Google Scholar] [CrossRef] [Green Version]
- Varsani, A.; Krupovic, M. Smacoviridae: A new family of animal-associated single-stranded DNA viruses. Arch. Virol. 2018, 163, 2005–2015. [Google Scholar] [CrossRef] [PubMed]
- Saikumar, G.; Das, T. Porcine circovirus. In Recent Advances in Animal Virology; Malik, Y.S., Singh, R.K., Yadav, M.P., Eds.; Springer: Singapore, 2019; pp. 171–195. [Google Scholar] [CrossRef]
- Cheung, A.K.; Ng, T.F.; Lager, K.M.; Bayles, D.O.; Alt, D.P.; Delwart, E.L.; Pogranichniy, R.M.; Kehrli, M.E., Jr. A divergent clade of circular single-stranded DNA viruses from pig feces. Arch. Virol. 2013, 158, 2157–2162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheung, A.K.; Ng, T.F.; Lager, K.M.; Alt, D.P.; Delwart, E.L.; Pogranichniy, R.M. Unique circovirus-like genome detected in pig feces. Genome Announc. 2014, 2, e00251-14. [Google Scholar] [CrossRef] [Green Version]
- Karlsson, O.E.; Larsson, J.; Hayer, J.; Berg, M.; Jacobson, M. The intestinal eukaryotic virome in healthy and diarrhoeic Neonatal Piglets. PLoS ONE 2016, 11, e0151481. [Google Scholar] [CrossRef] [Green Version]
- Kim, A.R.; Chung, H.C.; Kim, H.K.; Kim, E.O.; Nguyen, V.G.; Choi, M.G.; Yang, H.J.; Kim, J.A.; Park, B.K. Characterization of a complete genome of a circular single-stranded DNA virus from porcine stools in Korea. Virus Genes 2014, 48, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Oba, M.; Katayama, Y.; Naoi, Y.; Tsuchiaka, S.; Omatsu, T.; Okumura, A.; Nagai, M.; Mizutani, T. Discovery of fur seal feces-associated circular DNA virus in swine feces in Japan. J. Vet. Med. Sci. 2017, 79, 1664–1666. [Google Scholar] [CrossRef] [PubMed]
- Sachsenröder, J.; Twardziok, S.; Hammerl, J.A.; Janczyk, P.; Wrede, P.; Hertwig, S.; Johne, R. Simultaneous identification of DNA and RNA viruses present in pig faeces using process-controlled deep sequencing. PLoS ONE 2012, 7, e34631. [Google Scholar] [CrossRef]
- Shan, T.; Li, L.; Simmonds, P.; Wang, C.; Moeser, A.; Delwart, E. The fecal virome of pigs on a high-density farm. J. Virol. 2011, 85, 11697–11708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Z.; Liu, C.; Yang, H.; Chen, Y.; Liu, H.; Wei, L.; Liu, Z.; Jiang, Y.; He, X.; Wang, J. Fur seal feces-associated circular DNA virus identified in pigs in Anhui, China. Virol. Sin. 2021, 36, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Sikorski, A.; Arguello-Astorga, G.R.; Dayaram, A.; Dobson, R.C.; Varsani, A. Discovery of a novel circular single-stranded DNA virus from porcine faeces. Arch. Virol. 2013, 158, 283–289. [Google Scholar] [CrossRef]
- Sun, W.; Wang, W.; Cao, L.; Zheng, M.; Zhuang, X.; Zhang, H.; Yu, N.; Tian, M.; Lu, H.; Jin, N. Genetic characterization of three porcine circovirus-like viruses in pigs with diarrhoea in China. Transbound. Emerg. Dis. 2021, 68, 289–295. [Google Scholar] [CrossRef] [PubMed]
- Tochetto, C.; Varela, A.P.M.; Lima, D.A.; Loiko, M.R.; Scheffer, C.M.; Paim, W.P.; Cerva, C.; Schmidt, C.; Cibulski, S.P.; Ortiz, L.C.; et al. Viral DNA genomes in sera of farrowing sows with or without stillbirths. PLoS ONE 2020, 15, e0230714. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Tang, C.; Yue, H.; Ren, Y.; Song, Z. Viral metagenomics analysis demonstrates the diversity of viral flora in piglet diarrhoeic faeces in China. J. Gen. Virol. 2014, 95, 1603–1611. [Google Scholar] [CrossRef] [Green Version]
- Djikeng, A.; Halpin, R.; Kuzmickas, R.; Depasse, J.; Feldblyum, J.; Sengamalay, N.; Afonso, C.; Zhang, X.; Anderson, N.G.; Ghedin, E.; et al. Viral genome sequencing by random priming methods. BMC Genom. 2008, 9, 5. [Google Scholar] [CrossRef] [Green Version]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef] [PubMed]
- Huson, D.H.; Beier, S.; Flade, I.; Górska, A.; El-Hadidi, M.; Mitra, S.; Ruscheweyh, H.J.; Tappu, R. MEGAN community edition–interactive exploration and analysis of large-scale microbiome sequencing data. PLoS Comput. Biol. 2016, 12, e1004957. [Google Scholar] [CrossRef] [Green Version]
- Guindon, S.; Dufayard, J.F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [Green Version]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [Green Version]
- Martin, D.P.; Murrell, B.; Golden, M.; Khoosal, A.; Muhire, B. RDP4: Detection and analysis of recombination patterns in virus genomes. Virus Evol. 2015, 1, vev003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sikorski, A.; Dayaram, A.; Varsani, A. Identification of a novel circular DNA virus in New Zealand fur seal (Arctocephalus forsteri) fecal matter. Genome Announc. 2013, 1, e00558-13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Z.; He, Q.; Tang, C.; Zhang, B.; Yue, H. Identification and genomic characterization of a novel CRESS DNA virus from a calf with severe hemorrhagic enteritis in China. Virus Res. 2018, 255, 141–146. [Google Scholar] [CrossRef] [PubMed]
- Cheung, A.K. Porcine circovirus: Transcription and DNA replication. Virus Res. 2012, 164, 46–53. [Google Scholar] [CrossRef]
- Kaszab, E.; Lengyel, G.; Marton, S.; Dán, Á.; Bányai, K.; Fehér, E. Occurrence and genetic diversity of CRESS DNA viruses in wild birds: A Hungarian study. Sci. Rep. 2020, 10, 7036. [Google Scholar] [CrossRef]
- Nath, B.K.; Das, S.; Roby, J.A.; Sarker, S.; Luque, D.; Raidal, S.R.; Forwood, J.K. Structural perspectives of beak and feather disease virus and porcine circovirus proteins. Viral Immunol. 2021, 34, 49–59. [Google Scholar] [CrossRef] [PubMed]
- Blomström, A.; Fossum, C.; Wallgren, P.; Berg, M. Viral metagenomic analysis displays the co-infection situation in healthy and PMWS affected pigs. PLoS ONE 2016, 11, e0166863. [Google Scholar] [CrossRef]
- Kaszab, E.; Marton, S.; Forró, B.; Bali, K.; Lengyel, G.; Bányai, K.; Fehér, E. Characterization of the genomic sequence of a novel CRESS DNA virus identified in Eurasian jay (Garrulus glandarius). Arch. Virol. 2018, 163, 285–289. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Primer Name | Region | Sequenced Genome | Reference Strains | Annealing | Primer Sequences * |
|---|---|---|---|---|---|
| CVLV4-F CVLV4-R | ORF1 | 288_4 302_4 | po-circo-like virus 21 | 53 °C | 5’-ATCTTTGGTCTTGCATTGTTGC-3’ 5’-CTTCAAGGCTATCTTATCCTMCC-3’ |
| CVLV5-F CVLV5-R | rep | 302_5 303_5 453_5 | po-circo-like virus 41 | 58 °C | 5’- GACGGTTTTGACCCGTCAACAC-5’ 5’-CCACCACTTGTCAAACGGTTTGAAG-3’ |
| CVLV7-F CVLV7-R | ORF-2 | 303_7 453_7 | po-circo-like virus 51 | 53 °C | 5’-CTGCACCAATAGAAGATGGTAG-3’ 5’-GAGGTTCTGGAATTAAACCATTGTC-3’ |
| CVLV2-F CVLV2-R | cap | 306_2 451_2 | FSfaCVc | 53 °C | 5’-TAYCTTATGTGGACACATTTACCG-3’ 5’-TAAATTGTGGTTWGGACCATCC-3’ |
| Genome | Genome Length nt | Genome Type | Nonanucleotide Motif | ORF Position to the Nonanucleotide Motif | ||||
|---|---|---|---|---|---|---|---|---|
| rep | ORF1 | ORF2 | ORF3 | ORF4 | ||||
| 288_4 | 3926 | IV | CAGTATTAC | 1123–191 | 1232–1720 | 1733–2275 | 2289–2849 | 3249–2911 |
| 302_4 | 3926 | IV | CAGTATTAC | 1123–191 | 1232–1720 | 1733–2275 | 2289–2849 | 3249–2911 |
| 302_5 | 2946 | V | TAGTATTAC | 226–1203 | 1366–1941 | 1956–2543 | NA | NA |
| 303_5 | 2946 | V | TAGTATTAC | 226–1203 | 1366–1941 | 1956–2543 | NA | NA |
| 453_5 | 2942 | V | TAGTATTAC | 226–1203 | 1365–1940 | 1956–2543 | NA | NA |
| 303_7 | 2825 | V | CATTATTAC | 253–1212 | 1437–1964 | 1978–2583 | NA | NA |
| 453_7 | 2825 | V | CATTATTAC | 253–1212 | 1437–1964 | 1978–2583 | NA | NA |
| 306_2 | 2921 | II | TAGTATTAT | 2478–1423 | 14–1213 | 732–1112 | 2000–1509 | NA |
| 451_2 | 2912 | II | TAGTATTAT | 2469–1414 | 14–1204 | 723–1103 | 1991–1500 | NA |
| RCR Motifs | Superfamily 3 Helicase Motifs | |||||
|---|---|---|---|---|---|---|
| I | II | III | Walker-A | Walker-B | Motif C | |
| 288_4 | CFTIND | PHIQG | YCTK | GKGKS | VIDDW | ITSN |
| 302_4 | CFTIND | PHIQG | YCTK | GKGKS | VIDDW | ITSN |
| 302_5 | CFTINN | PHIQG | YCSK | GSGKT | VIDDY | VTSN |
| 303_5 | CFTINN | PHIQG | YCSK | GSGKT | VIDDY | VTSN |
| 453_5 | CFTINN | PHIQG | YCSK | GSGKT | VIDDY | VTSN |
| 303_7 | VFTINN | PHIQG | YCSK | GSGKT | LIDDF | ITSN |
| 453_7 | VFTINN | PHIQG | YCSK | GSGKT | LIDDF | ITSN |
| 306_2 | AMTVKN | QHCHI | YLAK | GSGKS | WFDEF | ISTI |
| 451_2 | ALTVKN | QHCHI | YLAK | GSGKS | WFDEF | ISTI |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fehér, E.; Mihalov-Kovács, E.; Kaszab, E.; Malik, Y.S.; Marton, S.; Bányai, K. Genomic Diversity of CRESS DNA Viruses in the Eukaryotic Virome of Swine Feces. Microorganisms 2021, 9, 1426. https://doi.org/10.3390/microorganisms9071426
Fehér E, Mihalov-Kovács E, Kaszab E, Malik YS, Marton S, Bányai K. Genomic Diversity of CRESS DNA Viruses in the Eukaryotic Virome of Swine Feces. Microorganisms. 2021; 9(7):1426. https://doi.org/10.3390/microorganisms9071426
Chicago/Turabian StyleFehér, Enikő, Eszter Mihalov-Kovács, Eszter Kaszab, Yashpal S. Malik, Szilvia Marton, and Krisztián Bányai. 2021. "Genomic Diversity of CRESS DNA Viruses in the Eukaryotic Virome of Swine Feces" Microorganisms 9, no. 7: 1426. https://doi.org/10.3390/microorganisms9071426