
Unrelated Fungal Rust Candidate Effectors Act on Overlapping Plant Functions

 ,
,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Growth conditions
2.2. RNA Extraction and Transcriptome Analysis
2.3. Metabolite Extraction and Metabolomics Analysis
2.4. Sequence Analysis and Integration
3. Results
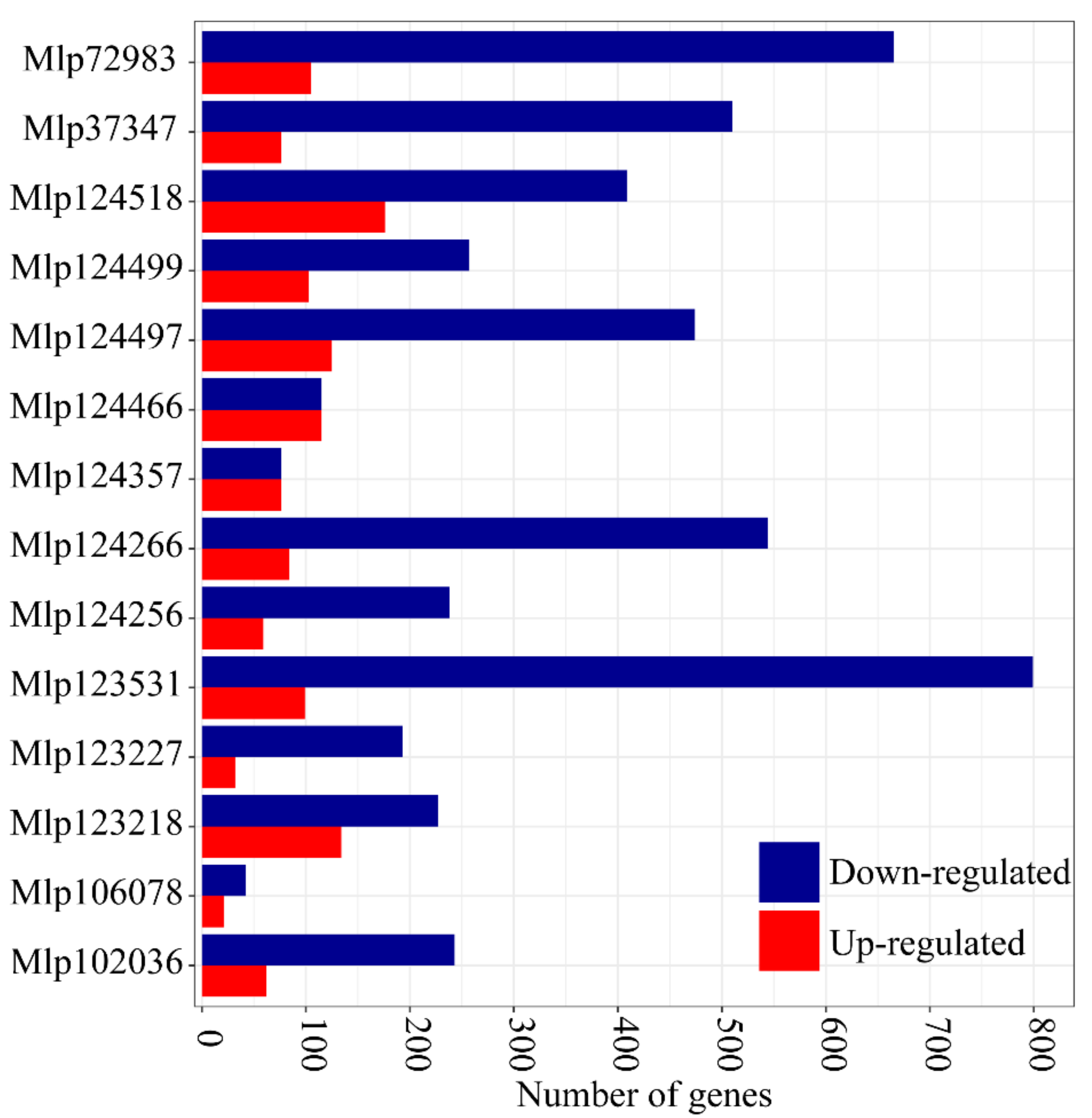
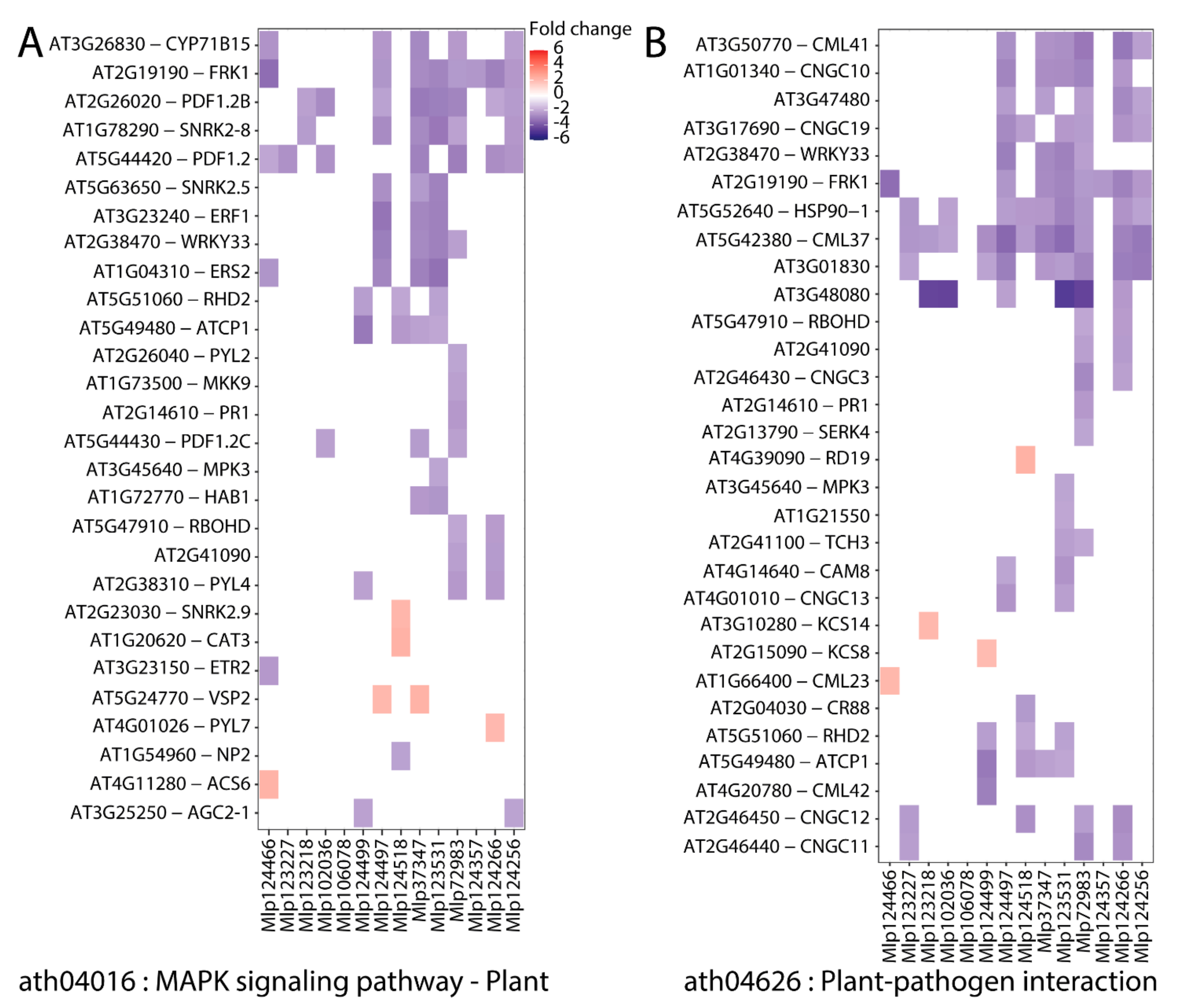
3.1. In Planta Expression of Candidate Fungal Effectors Results in Important Deregulation at the Transcriptome Level
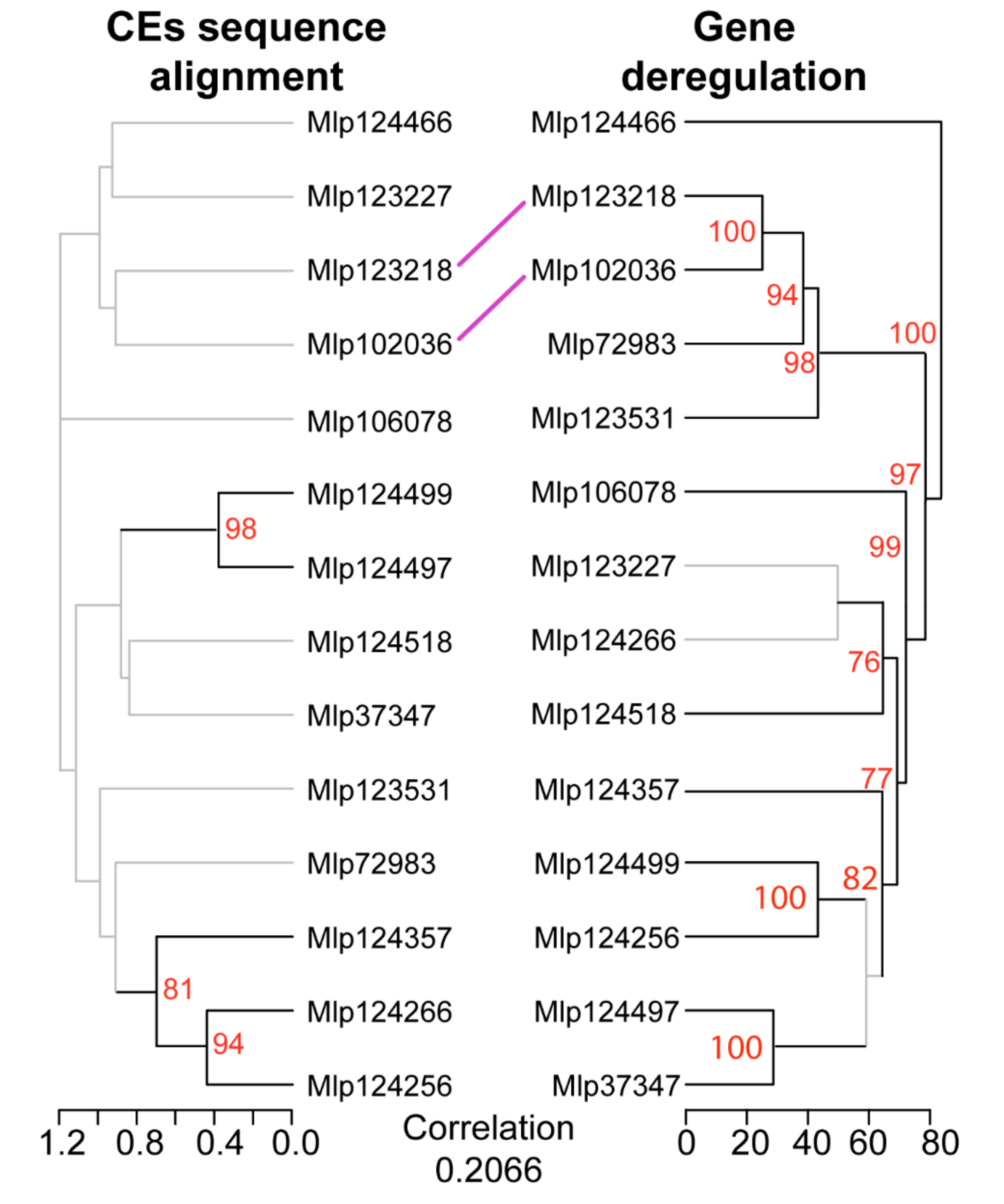
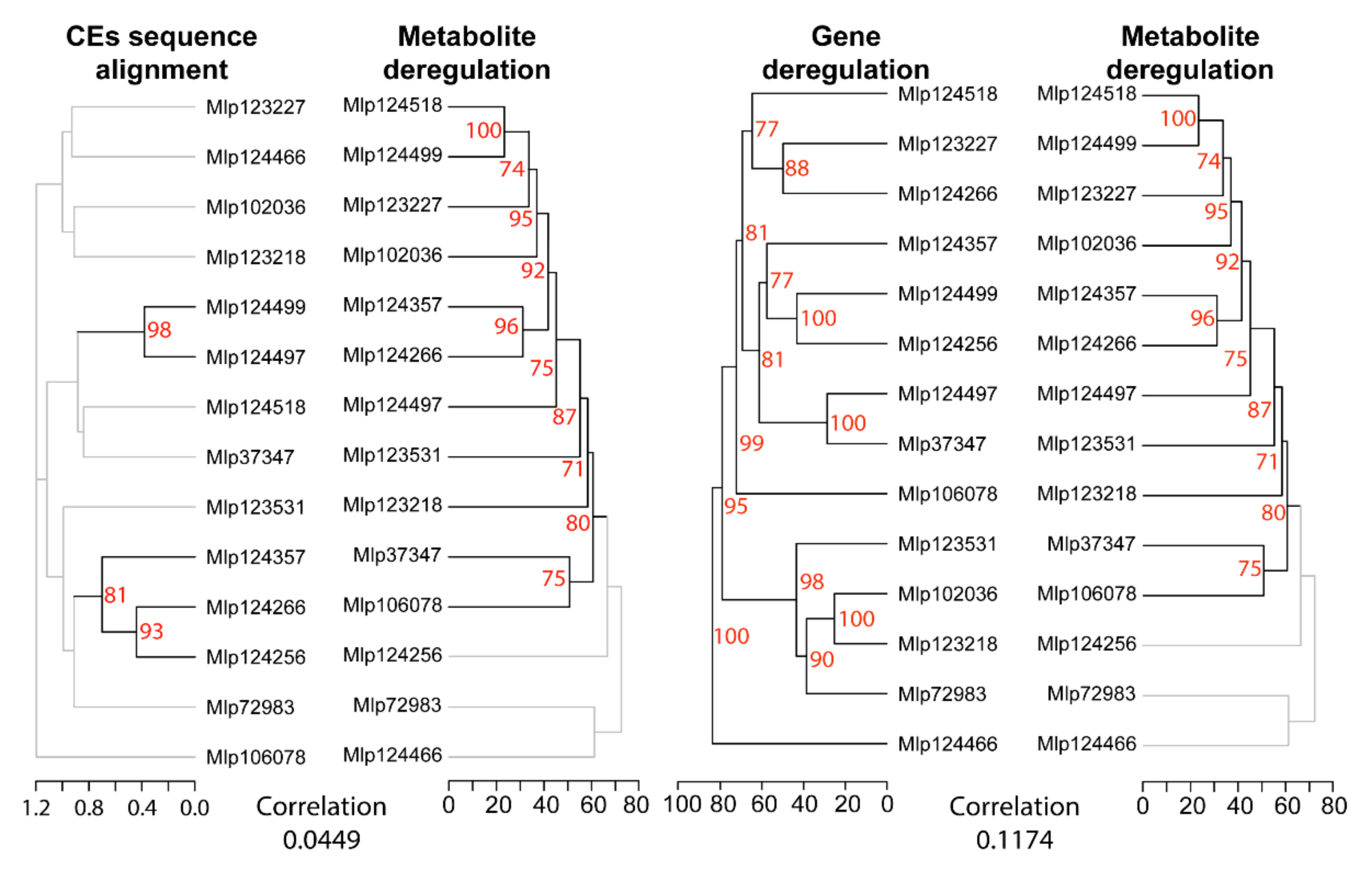
3.2. Hierarchical Clustering Based on Gene Expression Groups Effectors Independently of Amino Acid Sequence Homology
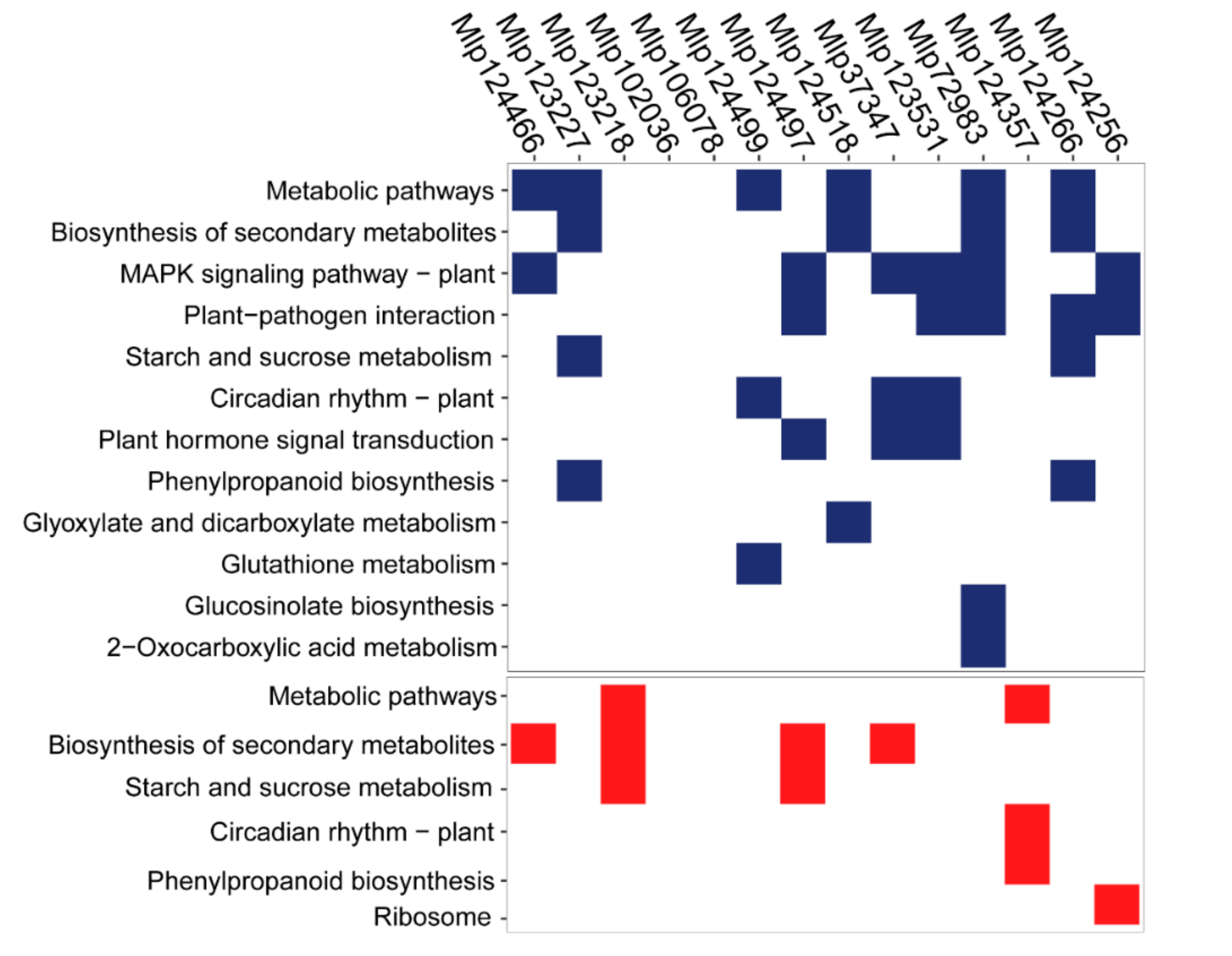
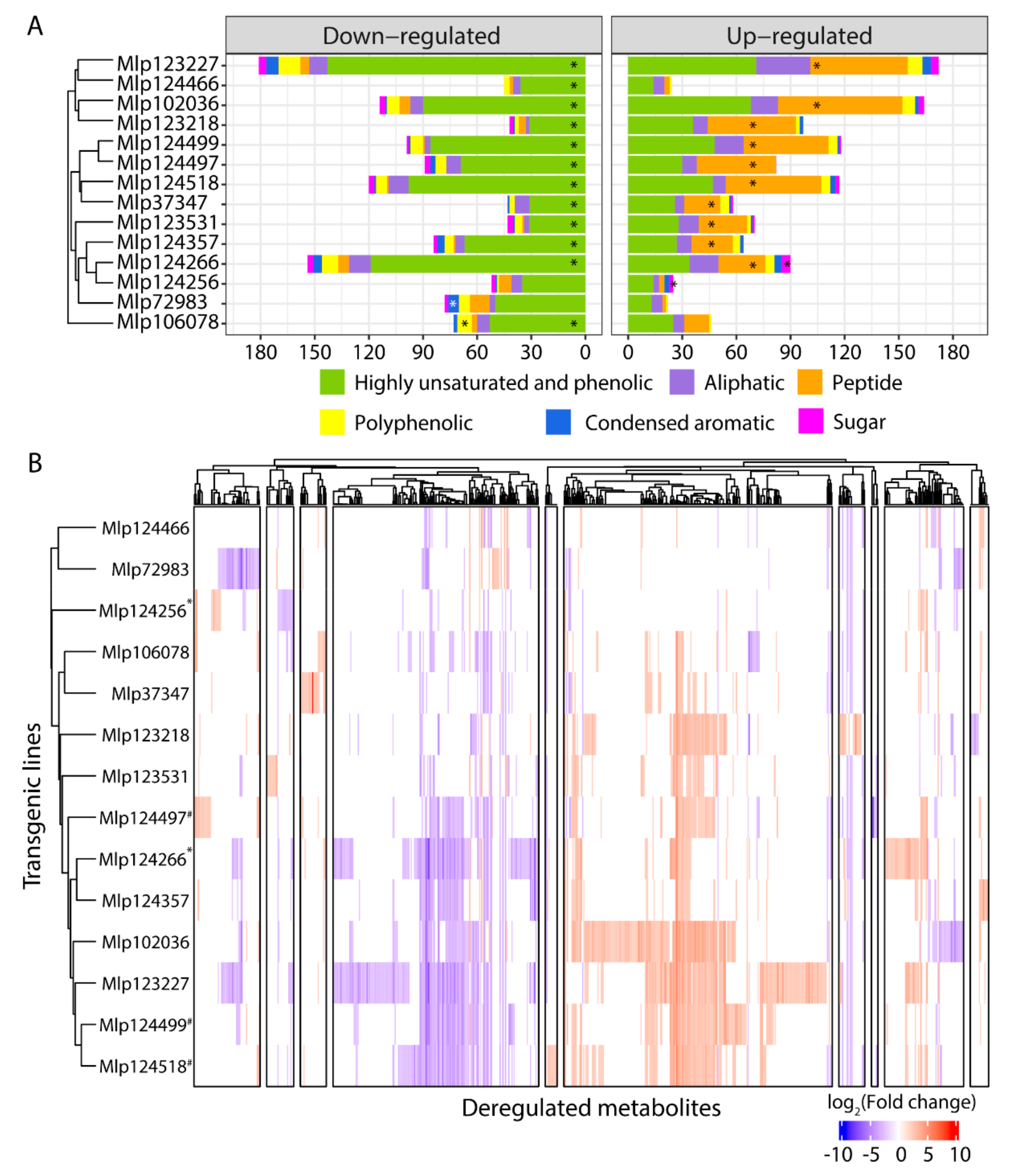
3.3. Effectors Converge on Deregulating the Same Metabolic Pathways while Others Display Unique Patterns
3.4. Similar to the Transcriptome, the Metabolome Is Deregulated by Several Effectors
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Henry, G.; Thonart, P.; Ongena, M. PAMPs, MAMPs, DAMPs and others: An update on the diversity of plant immunity elicitors. Biotechnol. Agron. Soc. Environ. 2012, 16, 257–268. [Google Scholar]
- Luna, E.; Pastor, V.; Robert, J.; Flors, V.; Mauch-Mani, B.; Ton, J. Callose deposition: A multifaceted plant defense response. Mol. Plant-Microbe Interact. 2011, 24, 183–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bigeard, J.; Colcombet, J.; Hirt, H. Signaling mechanisms in pattern-triggered immunity (PTI). Mol. Plant 2015, 8, 521–539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, J.D.G.; Dangl, J.L. The plant immune system. Nature 2006, 444, 323–329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dean, R.; Van Kan, J.A.; Pretorius, Z.A.; Hammond-Kosack, K.E.; Di Pietro, A.; Spanu, P.D.; Rudd, J.J.; Dickman, M.; Kahmann, R.; Ellis, J.; et al. The Top 10 fungal pathogens in molecular plant pathology. Mol. Plant Pathol. 2012, 13, 414–430. [Google Scholar] [CrossRef] [Green Version]
- Aime, M.C.; McTaggart, A.R.; Mondo, S.J.; Duplessis, S. Phylogenetics and phylogenomics of rust fungi. Adv. Genet. 2017, 100, 267–307. [Google Scholar] [CrossRef]
- Hogenhout, S.a.; Van der Hoorn, R.a.L.; Terauchi, R.; Kamoun, S. Emerging concepts in effector biology of plant-associated organisms. Mol. Plant-Microbe Interact. 2009, 22, 115–122. [Google Scholar] [CrossRef] [Green Version]
- Dangl, J.L.; Horvath, D.M.; Staskawicz, B.J. Pivoting the plant immune system from dissection to deployment. Science 2013, 341, 746–751. [Google Scholar] [CrossRef] [Green Version]
- Duplessis, S.; Cuomo, C.A.; Lin, Y.-C.; Aerts, A.; Tisserant, E.; Veneault-Fourrey, C.; Joly, D.L.; Hacquard, S.; Amselem, J.; Cantarel, B.L.; et al. Obligate biotrophy features unraveled by the genomic analysis of rust fungi. Proc. Natl. Acad. Sci. USA 2011, 108, 9166–9171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saunders, D.G.O.; Win, J.; Cano, L.M.; Szabo, L.J.; Kamoun, S.; Raffaele, S. Using hierarchical clustering of secreted protein families to classify and rank candidate effectors of rust fungi. PLoS ONE 2012, 7. [Google Scholar] [CrossRef]
- Enright, A.J.; Dongen, S.V.; Ouzounis, C.A. An efficient algorithm for large-scale detection of protein families. Nucleic Acids Res. 2002, 30, 1575–1584. [Google Scholar] [CrossRef] [PubMed]
- Ravensdale, M.; Nemri, A.; Thrall, P.H.; Ellis, J.G.; Dodds, P.N. Co-evolutionary interactions between host resistance and pathogen effector genes in flax rust disease. Mol. Plant Pathol. 2010, 12, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Sperschneider, J.; Dodds, P.N.; Gardiner, D.M.; Manners, J.M.; Singh, K.B.; Taylor, J.M. Advances and challenges in computational prediction of effectors from plant pathogenic fungi. PLoS Pathog. 2015, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lorrain, C.; Hecker, A.; Duplessis, S. Effector-mining in the poplar rust fungus Melampsora larici-populina. Front. Plant Sci. 2015, 6. [Google Scholar] [CrossRef] [Green Version]
- Lyu, X.; Shen, C.; Fu, Y.; Xie, J.; Jiang, D.; Li, G.; Cheng, J. A small secreted virulence-related protein isessential for the necrotrophic interactions of Sclerotinia sclerotiorum with its host plants. PLoS Pathog. 2016, 12. [Google Scholar] [CrossRef]
- Li, Z.; Yin, Z.; Fan, Y.; Xu, M.; Kang, Z.; Huang, L. Candidate effector proteins of the necrotrophic apple canker pathogen Valsa mali can suppress BAX-induced PCD. Front. Plant Sci. 2015, 6, 579. [Google Scholar] [CrossRef] [Green Version]
- Lorrain, C.; Petre, B.; Duplessis, S. Show me the way: Rust effector targets in heterologous plant systems. Curr. Opin. Microbiol. 2018, 46, 19–25. [Google Scholar] [CrossRef]
- Chaudhari, P.; Ahmed, B.; Joly, D.L.; Germain, H. Effector biology during biotrophic invasion of plant cells. Virulence 2014, 5, 703–709. [Google Scholar] [CrossRef] [Green Version]
- Pitino, M.; Armstrong, C.M.; Cano, L.M.; Duan, Y. Transient expression of Candidatus Liberibacter Asiaticus effector induces cell death in Nicotiana benthamiana. Front. Plant Sci. 2016, 7. [Google Scholar] [CrossRef]
- Jamir, Y.; Guo, M.; Oh, H.S.; Petnicki-Ocwieja, T.; Chen, S.; Tang, X.; Dickman, M.B.; Collmer, A.; Alfano, J.R. Identification of Pseudomonas syringae type III effectors that can suppress programmed cell death in plants and yeast. Plant J. 2004, 37, 554–565. [Google Scholar] [CrossRef]
- Houterman, P.M.; Ma, L.; van Ooijen, G.; De Vroomen, M.J.; Cornelissen, B.J.C.; Takken, F.L.W.; Rep, M. The effector protein Avr2 of the xylem-colonizing fungus Fusarium oxysporum activates the tomato resistance protein I-2 intracellularly. Plant J. 2009, 58, 970–978. [Google Scholar] [CrossRef]
- Germain, H.; Joly, D.L.; Mireault, C.; Plourde, M.B.; Letanneur, C.; Stewart, D.; Morency, M.-J.; Petre, B.; Duplessis, S.; Séguin, A. Infection assays in Arabidopsis reveal candidate effectors from the poplar rust fungus that promote susceptibility to bacteria and oomycete pathogens. Mol. Plant Pathol. 2018, 19, 191–200. [Google Scholar] [CrossRef] [Green Version]
- Bentem, S.D.L.F.v.; Vossen, J.H.; Vries, K.J.d.; Wees, S.v.; Tameling, W.I.L.; Dekker, H.L.; Koster, C.G.d.; Haring, M.A.; Takken, F.L.W.; Cornelissen, B.J.C. Heat shock protein 90 and its co-chaperone protein phosphatase 5 interact with distinct regions of the tomato I-2 disease resistance protein. Plant J. 2005, 43, 284–298. [Google Scholar] [CrossRef]
- Ahmed, M.B.; Santos, K.C.G.d.; Petre, B.; Lorrain, C.; Duplessis, S.; Desgagne-Penix, I.; Germain, H. A rust fungal effector binds plant DNA and modulates transcription. Nat. Sci. Rep. 2018, 8. [Google Scholar] [CrossRef] [PubMed]
- Kunjeti, S.G.; Iyer, G.; Johnson, E.; Li, E.; Broglie, K.E. Identification of Phakopsora pachyrhizi candidate effectors with virulence activity in a distantly related pathosystem. Front. Plant Sci. 2016, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caillaud, M.C.; Piquerez, S.J.M.; Fabro, G.; Steinbrenner, J.; Ishaque, N.; Beynon, J.; Jones, J.D.G. Subcellular localization of the Hpa RxLR effector repertoire identifies a tonoplast-associated protein HaRxL17 that confers enhanced plant susceptibility. Plant J. 2012, 69, 252–265. [Google Scholar] [CrossRef]
- Petre, B.; Saunders, D.G.O.; Sklenar, J.; Lorrain, C.; Win, J.; Duplessis, S.; Kamoun, S. Candidate effector proteins of the rust pathogen Melampsora larici-populina target diverse plant cell compartments. Mol. Plant-Microbe Interact. 2015, 28, 689–700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petre, B.; Saunders, D.G.O.; Sklenar, J.; Lorrain, C.; Krasileva, K.V.; Win, J.; Duplessis, S.; Kamoun, S. Heterologous expression screens in Nicotiana benthamiana identify a candidate effector of the wheat yellow rust pathogen that associates with processing bodies. PLoS ONE 2016, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plett, J.M.; Kemppainen, M.; Kale, S.D.; Kohler, A.; Legué, V.; Brun, A.; Tyler, B.M.; Pardo, A.G.; Martin, F. A secreted effector protein of Laccaria bicolor is required for symbiosis development. Curr. Biol. 2011, 21, 1197–1203. [Google Scholar] [CrossRef] [Green Version]
- Madina, M.H.; Rahman, M.S.; Huang, X.; Zhang, Y.; Zheng, H.; Germain, H. A poplar rust effector protein associates with protein disulfide isomerase and enhances plant susceptibility. Biology 2020, 9, 294. [Google Scholar] [CrossRef]
- Amack, S.C.; Antunes, M.S. CaMV35S promoter—A plant biology and biotechnology workhorse in the era of synthetic biology. Curr. Plant Biol. 2020, 24. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef] [PubMed]
- Morgan, M.; Pagès, H.; Obenchain, V.; Hayden, N. Bioconductor—Rsamtools: Binary Alignment (BAM), FASTA, Variant Call (BCF), and Tabix File Import. R Package Version 2.2.3. Available online: http://bioconductor.riken.jp/packages/3.10/bioc/html/Rsamtools.html (accessed on 27 February 2019).
- Lawrence, M.; Huber, W.; Pagès, H.; Aboyoun, P.; Carlson, M.; Gentleman, R.; Morgan, M.; Carey, V. Software for Computing and Annotating Genomic Ranges. PLoS Compu. Biol. 2013, 9. [Google Scholar] [CrossRef]
- Dos Santos, K.C.G.; Desgagne-Penix, I.; Germain, H. Custom selected reference genes outperform pre-defined reference genes in transcriptomic analysis. BMC Genom. 2020, 21. [Google Scholar] [CrossRef] [PubMed]
- Dos Santos, K.C.G.; Pelletier, G.; Séguin, A.; Guillemette, F.; Hawkes, J.A.; Desgagné-Penix, I.; Germain, H. FigShare—Supplementary Material: Differential Alteration of Plant Functions by Homologous Fungal Candidate Effectors. Available online: https://doi.org/10.6084/m9.figshare.13166501.v3 (accessed on 29 October 2020).
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Yu, G.; Wang, L.; Han, Y.; He, Q. clusterProfiler: An R package for comparing biological themes among gene clusters. OMICS J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Zhao, S.; Guo, Y.; Shyr, Y. Bioconductor — KEGGprofile: An Annotation and Visualization Package for Multi-Types and Multi-Groups Expression Data in KEGG Pathway. R Package Version 1.24.0. Available online: https://bioconductor.org/packages/3.8/bioc/html/KEGGprofile.html (accessed on 27 February 2019).
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, R.; Terada, Y.; Shimodaira, H. Github—Pvclust: Hierarchical Clustering with p-Values via Multiscale Bootstrap Resampling. R Package Version 2.2-0. Available online: https://github.com/shimo-lab/pvclust (accessed on 11 January 2020).
- Hawkes, J.A.; Patriarca, C.; Sjöberg, P.J.R.; Tranvik, L.J.; Bergquist, J. Extreme isomeric complexity of dissolved organic matter found across aquatic environments. Limnol. Oceanogr. Lett. 2018, 3, 21–30. [Google Scholar] [CrossRef] [Green Version]
- Koch, B.P.; Dittmar, T. From mass to structure: An aromaticity index for high-resolution mass data of natural organic matter. Rapid Commun. Mass Spectrom. 2006, 20, 926–932. [Google Scholar] [CrossRef] [Green Version]
- Kellerman, A.M.; Guillemette, F.; Podgorski, D.C.; Aiken, G.R.; Butler, K.D.; Spencer, R.G.M. Unifying concepts linking dissolved organic matter composition to persistence in aquatic ecosystems. Environ. Sci. Technol. 2018, 52, 2538–2548. [Google Scholar] [CrossRef] [PubMed]
- Leader, D.P.; Burgess, K.; Creek, D.; Barrett, M.P. Pathos: A web facility that uses metabolic maps to display experimental changes in metabolites identified by mass spectrometry. Rapid Commun. Mass Spectrom. 2011, 25, 3422–3426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 37, 1237–1239. [Google Scholar] [CrossRef] [PubMed]
- Madeira, F.; Park, Y.M.; Lee, J.; Buso, N.; Gur, T.; Madhusoodanan, N.; Basutkar, P.; Tivey, A.R.N.; Potter, S.C.; Finn, R.D.; et al. The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Res. 2019, 47, W636–W641. [Google Scholar] [CrossRef] [Green Version]
- Galili, T. dendextend: An R package for visualizing, adjusting, and comparing trees of hierarchical clustering. Bioinformatics 2015, 31, 3718–3720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaouar, O.; Morency, M.-J.; Letanneur, C.; Séguin, A.; Germain, H. The 124202 candidate effector of Melampsora larici-populina interacts with membranes in Nicotiana and Arabidopsis. Can. J. Plant Pathol. 2016, 38, 197–208. [Google Scholar] [CrossRef]
- Duplessis, S.; Hacquard, S.; Delaruelle, C.; Tisserant, E.; Frey, P.; Martin, F.; Kohler, A. Melampsora larici-populina transcript profiling during germination and timecourse infection of poplar leaves reveals dynamic expression patterns associated with virulence and biotrophy. Mol. Plant-Microbe Interact. 2011, 24, 808–818. [Google Scholar] [CrossRef] [Green Version]
- Dodds, P.N.; Lawrence, G.J.; Catanzariti, A.-M.; Ayliffe, M.A.; Ellis, J.G. The Melampsora lini AvrL567 Avirulence genes are expressed in haustoria and their products are recognized inside plant cells. Plant Cell 2004, 16, 755–768. [Google Scholar] [CrossRef] [Green Version]
- Gan, P.H.P.; Rafiqi, M.; Ellis, J.G.; Jones, D.A.; Hardham, A.R.; Dodds, P.N. Lipid binding activities of flax rust AvrM and AvrL567 effectors. Plant Signal. Behav. 2010, 5, 1272–1275. [Google Scholar] [CrossRef] [Green Version]
- Lorrain, C.; Marchal, C.; Hacquard, S.; Delaruelle, C.; Péytowski, J.; Petre, B.; Hecker, A.; Frey, P.; Duplessis, S. The rust fungus Melampsora larici-populina expresses a conserved genetic program and distinct sets of secreted protein genes during infection of its two host plants, larch and poplar. Mol. Plant-Microbe Interact. 2018, 31, 695–706. [Google Scholar] [CrossRef] [Green Version]
- Dos Santos, K.C.G.; Pelletier, G.; Séguin, A.; Guillemette, F.; Hawkes, J.A.; Desgagné-Penix, I.; Germain, H. Embl Metabolights—MTBLS2096: Differential Alteration of Plant Functions by Homologous Fungal Candidate Effectors. Available online: https://www.ebi.ac.uk/metabolights/MTBLS2096/descriptors (accessed on 3 December 2020).
- Selin, C.; Kievit, T.R.d.; Belmonte, M.F.; Fernando, W.G.D. Elucidating the role of effectors in plant-fungal interactions: Progress and challenges. Front. Microbiol. 2016, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, S.M. Identification and Functional Characterization of Powdery Mildew Effectors. Doctoral Dissertation, Universität zu Köln, Köln, Germany, 2009. [Google Scholar]
- Ramachandran, S.R.; Yin, C.; Kud, J.; Tanaka, K.; Mahoney, A.K.; Xiao, F.; Hulbert, S.H. Effectors from wheat rust fungi suppress multiple plant defense responses. Phytopathology 2016, 107, 75–83. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Yang, Y.; Wang, C.; Liu, M.; Li, H.; Fu, Y.; Wang, Y.; Nie, Y.; Liu, X.; Ji, W. Large-scale transcriptome comparison reveals distinct gene activations in wheat responding to stripe rust and powdery mildew. BMC Genom. 2014, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trujillo-Moya, C.; Ganthaler, A.; Stöggl, W.; Kranner, I.; Schüler, S.; Ertl, R.; Schlosser, S.; George, J.-P.; Mayr, S. RNA-Seq and secondary metabolite analyses reveal a putative defence-transcriptome in Norway spruce (Picea abies) against needle bladder rust (Chrysomyxa rhododendri) infection. BMC Genom. 2020, 21. [Google Scholar] [CrossRef]
- Tremblay, A.; Hosseini, P.; Alkharouf, N.W.; Li, S.; Matthews, B.F. Transcriptome analysis of a compatible response by Glycine max to Phakopsora pachyrhizi infection. Plant Sci. 2010, 179, 183–193. [Google Scholar] [CrossRef]
- Tao, S.; Auer, L.; Morin, E.; Liang, Y.-M.; Duplessis, S. Transcriptome analysis of apple leaves infected by the rust fungus Gymnosporangium yamadae at two sporulation stages reveals detoxication and secondary metabolite host responses and fungal pathogenesis related genes. Mol. Plant-Microbe Interact. 2020, 33, 444–461. [Google Scholar] [CrossRef]
- Chandra, S.; Singh, D.; Pathak, J.; Kumari, S.; Kumar, M.; Poddar, R.; Balyan, H.S.; Gupta, P.K.; Prabhu, K.V.; Mukhopadhyay, K. De novo assembled wheat transcriptomes delineate differentially expressed host genes in response to leaf rust infection. PLoS ONE 2016, 11. [Google Scholar] [CrossRef] [PubMed]
- Azaiez, A.; Boyle, B.; Levée, V.; Séguin, A. Transcriptome profiling in hybrid poplar following interactions with Melampsora rust fungi. Mol. Plant-Microbe Interact. 2009, 22, 190–200. [Google Scholar] [CrossRef] [PubMed]
- Meng, Q.; Gupta, R.; Kwon, S.J.; Wang, Y.; Agrawal, G.K.; Rakwal, R.; Park, S.R.; Kim, S.T. Transcriptomic analysis of Oryza sativa leaves reveals key changes in response to Magnaporthe oryzae MSP1. Plant Pathol. J. 2018, 34, 257–268. [Google Scholar] [CrossRef]
- Hacquard, S.; Joly, D.L.; Lin, Y.C.; Tisserant, E.; Feau, N.; Delaruelle, C.; Legué, V.; Kohler, A.; Tanguay, P.; Petre, B.; et al. A comprehensive analysis of genes encoding small secreted proteins identifies candidate effectors in Melampsora larici-populina (poplar leaf rust). Mol. Plant-Microbe Interact. 2012, 25, 279–293. [Google Scholar] [CrossRef] [Green Version]
- Joshi, T.; Xu, D. Quantitative assessment of relationship between sequence similarity and function similarity. BMC Genom. 2007, 8. [Google Scholar] [CrossRef] [Green Version]
- Haas, B.J.; Kamoun, S.; Zody, M.C.; Jiang, R.H.Y.; Handsaker, R.E.; Cano, L.M.; Grabherr, M.; Kodira, C.D.; Raffaele, S.; Torto-Alalibo, T.; et al. Genome sequence and analysis of the Irish potato famine pathogen Phytophthora infestans. Nature 2009, 461, 393–398. [Google Scholar] [CrossRef] [PubMed]
- Evangelisti, E.; Gogleva, A.; Hainaux, T.; Doumane, M.; Tulin, F.; Quan, C.; Yunusov, T.; Floch, K.; Schornack, S. Time-resolved dual transcriptomics reveal early induced Nicotiana benthamiana root genes and conserved infection-promoting Phytophthora palmivora effectors. BMC Biol. 2017, 15, 39. [Google Scholar] [CrossRef]
- Anderson, J.P.; Sperschneider, J.; Win, J.; Kidd, B.; Yoshida, K.; Hane, J.; Saunders, D.G.O.; Singh, K.B. Comparative secretome analysis of Rhizoctonia solani isolates with different host ranges reveals unique secretomes and cell death inducing effectors. Nat. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Castañeda-Ojeda, M.P.; López-Solanilla, E.; Ramos, C. Differential modulation of plant immune responses by diverse members of the Pseudomonas savastanoi pv. savastanoi HopAF type III effector family. Mol. Plant Pathol. 2017, 18, 625–634. [Google Scholar] [CrossRef]
- Remigi, P.; Anisimova, M.; Guidot, A.; Genin, S.; Peeters, N. Functional diversification of the GALA type III effector family contributes to Ralstonia solanacearum adaptation on different plant hosts. New Phytol. 2011, 192, 976–987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.-G.; Taylor, K.W.; Mudgett, M.B. Comparative analysis of the XopD type III secretion (T3S) effector family in plant pathogenic bacteria. Mol. Plant Pathol. 2011, 12, 715–730. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, S.; Schweizer, G.; Rössel, N.; Thines, M.; Kahmann, R. Neofunctionalization of the secreted Tin2 effector in the fungal pathogen Ustilago maydis. Nat. Microbiol. 2019, 4, 251–257. [Google Scholar] [CrossRef] [PubMed]
- Zenser, N.; Ellsmore, A.; Leasure, C.; Callis, J. Auxin modulates the degradation rate of Aux/IAA proteins. Proc. Natl. Acad. Sci. USA 2001, 98, 11795–11800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iglesias, M.J.; Terrile, M.C.; Casalongué, C.A. Auxin and salicylic acid signalings counteract the regulation of adaptive responses to stress. Plant Signal. Behav. 2011, 6, 452–454. [Google Scholar] [CrossRef]
- Popa, C.; Coll, N.S.; Valls, M.; Sessa, G. Yeast as a heterologous model system to uncover type III effector function. PLoS Pathog. 2016, 12. [Google Scholar] [CrossRef] [Green Version]
- Rice, D.W.; Sheehan, K.B.; Newton, I.L.G. Large-scale identification of Wolbachia pipientis effectors. Genome Biol. Evol. 2017, 9, 1925–1937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cunha, M.d.; Milho, C.; Almeida, F.; Pais, S.V.; Borges, V.; Maurício, R.; Borrego, M.J.; Gomes, J.P.; Mota, L.J. Identification of type III secretion substrates of Chlamydia trachomatis using Yersinia enterocolitica as a heterologous system. BMC Microbiol. 2014, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CE | Length (Cysteine) | Family (Members) | Subcellular Localization a | U, P, B, L b,c |
|---|---|---|---|---|
| Mlp37347 | 151 (2) | - | Plasmodesmata | E, HE, E, E |
| Mlp72983 | 220 (8) | CPG332-CPG333(13) | Chloroplast | E, HE, E, HE |
| Mlp102036 | 107 (0) | CPG2528(5) | Nucleocytosolic | E, HE, E, E |
| Mlp106078 | 137 (10) | - | Nucleocytosolic | E, HE, E, E |
| Mlp123218 | 209 (6) | CPG543(7) | Nucleocytosolic | E, HE, E, E |
| Mlp123227 | 124 (3) | CPG1059(2) | Nucleocytosolic | E, HE, E, HE |
| Mlp123531 | 102 (8) | CPG4557(3) | Nucleocytosolic | E, HE, E, E |
| Mlp124256 | 89 (6) | CPG5464(13) | Nucleocytosolic | N, N, E, E |
| Mlp124266 | 92 (7) | CPG5464(13) | Nucleocytosolic | N, N, E, E |
| Mlp124357 | 98 (6) | CPG4890 | Tonoplast | N, N, E, E |
| Mlp124466 | 76 (0) | - | Nucleocytosolic | - |
| Mlp124497 | 77 (4) | CPGH1(33) | Nucleocytosolic | N, N, N, N |
| Mlp124499 | 72 (3) | CPGH1(33) | Nucleocytosolic | N, N, E, HE |
| Mlp124518 | 76 (3) | CPGH1(33) | Nucleocytosolic | N, N, E, E |
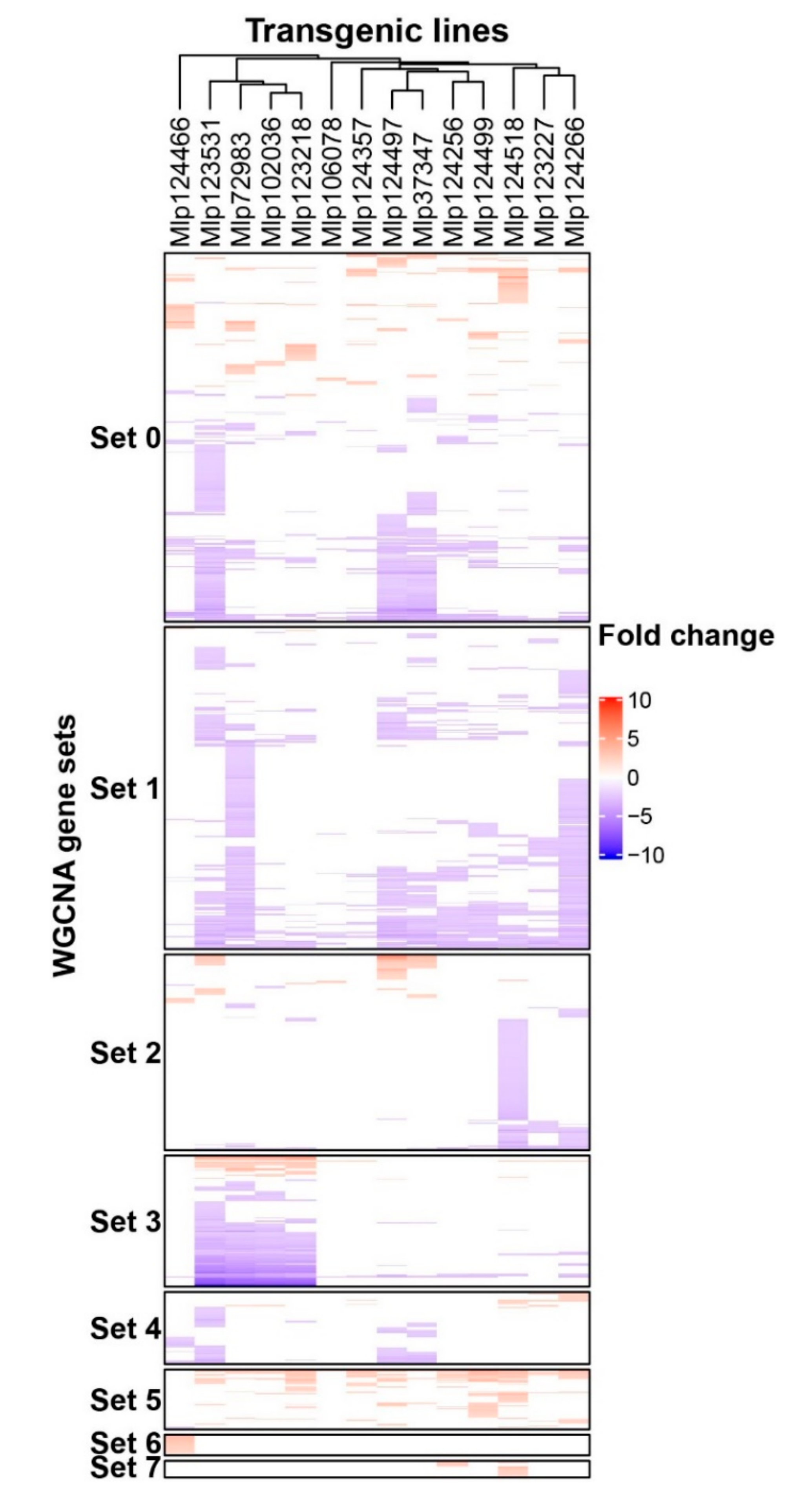
| Set | Genes in the Set | Up-Regulated a | Down-Regulated a | Enriched GO Terms |
|---|---|---|---|---|
| Set 0 | 714 | 262 | 451 | Response to water deprivation |
| Cold acclimation; Leaf senescence | ||||
| Response to fungus, to chitin, to ROS | ||||
| Response to salt stress and to hypoxia | ||||
| Defense response to fungus Response to toxic substance | ||||
| Response to nitrogen compound and to ET | ||||
| Isoprenoid, triterpenoid and terpenoid biosynthesis | ||||
| Plant-type cell wall loosening | ||||
| Phosphorelay signal transduction system | ||||
| Set 1 | 624 | 10 | 615 | Response to drug, nitrogen, ROS and ozone |
| Response to SA, JA and karrikin | ||||
| Response to wounding, to herbivore and insect | ||||
| Cellular response to light stimulus and hypoxia | ||||
| Cellular response to acid chemical | ||||
| Defense response (incompatible interaction) | ||||
| Defense response by callose deposition in cell wall | ||||
| Defense response by cell wall thickening | ||||
| SAR and ISR | ||||
| Camalexin, indole phytoalexin and SA biosynthesis | ||||
| Sulfur compound biosynthesis | ||||
| Toxin and phenol-containing compound biosynthesis | ||||
| Set 2 | 379 | 89 | 290 | Response to karrikin, to nutrient levels and to copper ion |
| S-glycoside and unsaturated fatty acid biosynthesis | ||||
| Chlorophyll biosynthesis | ||||
| Tetraterpenoid, terpenoid and carotenoid biosynthesis | ||||
| Isoprenoid, glycosyl and xanthophyll metabolism | ||||
| Sulfur compound, cofactor and leucine biosynthesis | ||||
| Defense response to insect | ||||
| De-etiolation; Chloroplast organization | ||||
| Set 3 | 253 | 47 | 207 | No GO term enriched |
| Set 4 | 140 | 32 | 109 | Response to water deprivation |
| Response to salt stress and to starvation | ||||
| Cellular amino acid catabolism/metabolism | ||||
| ET-activated signaling pathway | ||||
| Indole-containing compound metabolism | ||||
| Set 5 | 116 | 113 | 4 | Circadian rhythm; Starch catabolism |
| Response to cold | ||||
| Regulation of reproductive process | ||||
| Regulation of post-embryonic development | ||||
| Set 6 | 40 | 38 | 2 | Response to hypoxia and to wounding |
| Response to drug, to chitin and to salt stress | ||||
| Transcription; Phloem or xylem histogenesis | ||||
| Set 7 | 32 | 32 | 0 | Photosynthesis; Proton transmembrane transport |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
dos Santos, K.C.G.; Pelletier, G.; Séguin, A.; Guillemette, F.; Hawkes, J.; Desgagné-Penix, I.; Germain, H. Unrelated Fungal Rust Candidate Effectors Act on Overlapping Plant Functions. Microorganisms 2021, 9, 996. https://doi.org/10.3390/microorganisms9050996
dos Santos KCG, Pelletier G, Séguin A, Guillemette F, Hawkes J, Desgagné-Penix I, Germain H. Unrelated Fungal Rust Candidate Effectors Act on Overlapping Plant Functions. Microorganisms. 2021; 9(5):996. https://doi.org/10.3390/microorganisms9050996
Chicago/Turabian Styledos Santos, Karen Cristine Goncalves, Gervais Pelletier, Armand Séguin, François Guillemette, Jeffrey Hawkes, Isabel Desgagné-Penix, and Hugo Germain. 2021. "Unrelated Fungal Rust Candidate Effectors Act on Overlapping Plant Functions" Microorganisms 9, no. 5: 996. https://doi.org/10.3390/microorganisms9050996