
Gut Microbiota and NAFLD: Pathogenetic Mechanisms, Microbiota Signatures, and Therapeutic Interventions

 , , , ,
, , , ,
Abstract
:1. Introduction
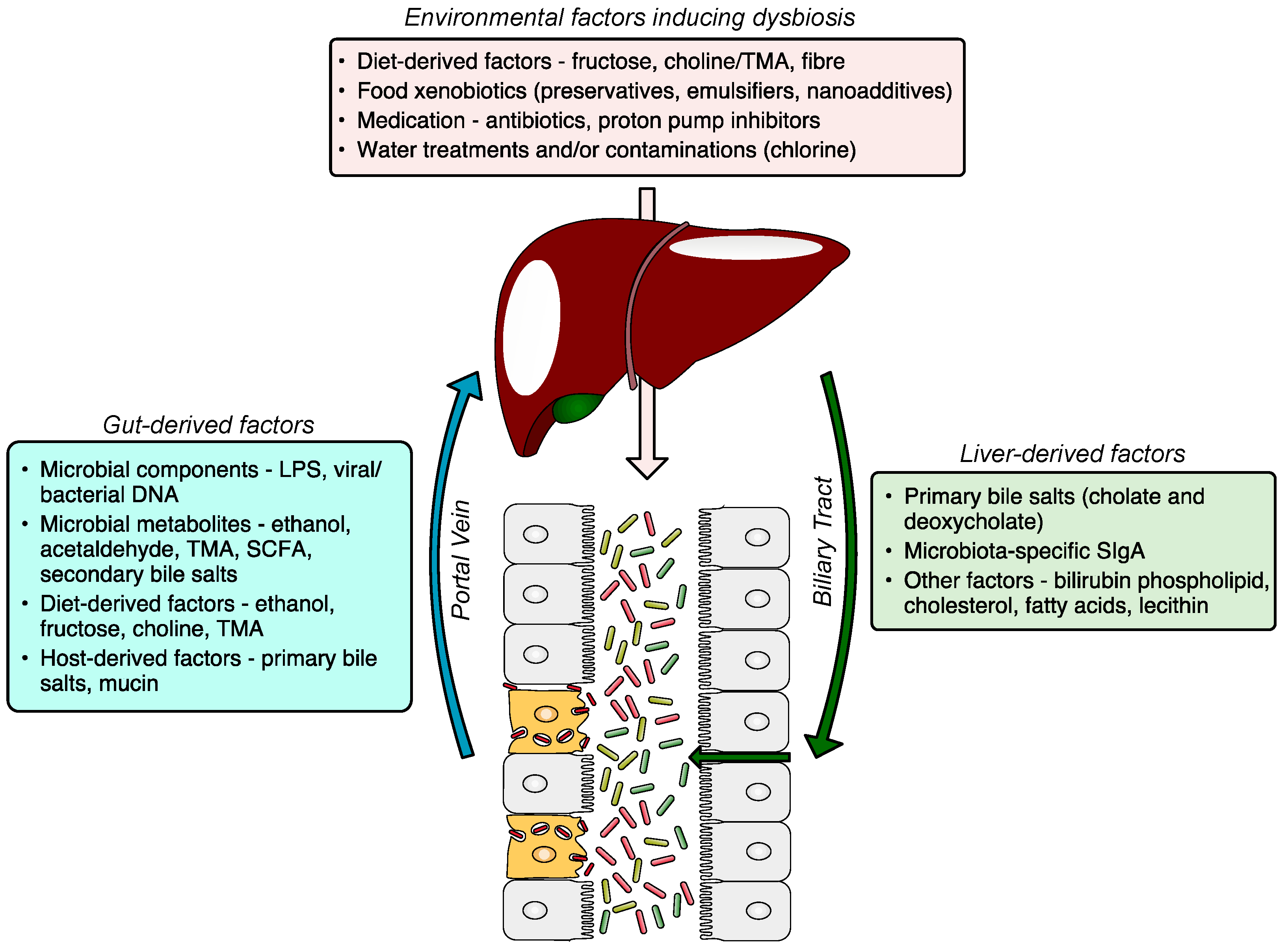
2. Gut Microbiota Dysbiosis
2.1. Introduction to Dysbiosis
2.2. Triggers and Drivers of Dysbiosis
2.3. Consequences of Dysbiosis
3. NAFLD-Associated Microbiota Signatures
3.1. Gut Microbiota Signatures

{kind=link}
| Phylum | Class | Family | Genus |
|---|---|---|---|
| Proteobacteria↑ [11,12,30,38,39] | Gammaproteobacteria↑ [40] | Enterobacteriaceae↑ [11,12] | Shigella↑ [11] |
| Escherichia↑ [12,30,38] | |||
| Pasteurellaceae↑ [39] | Haemophilus↓ [38] | ||
| Succinivibrionaceae↑ [41] | |||
| Epsilonproteobacteria↑ [40] | |||
| Alphaproteobacteria | Kiloniellaceae↑ [39] | ||
| Bradyrhizobiaceae | Bradyrhizobium↑ [42] | ||
| Verrucomicrobia↑ [38] | Verrucomicrobiae | Akkermansiaceae | Akkermansia↑ [38] |
| Fusobacteria↑ [11] | |||
| Bacteroidetes↑↓ [11,12,40,42,43,44] | Bacteroidia↑ [43] | Rikenellaceae↓ [12,42] | Alistipes↓ [12] |
| Bacteroidaceae | Bacteroides↑ [45] | ||
| Bacteroidetes | Prevotellaceae↑↓ [11,12] | Prevotella↑↓ [11,12,40,45] | |
| Porphyromonadaceae↑↓ [39,43] | Porphyromonas↑ [12] | ||
| Parabacteroides↑ [41] | |||
| Coprobacter↓ [38] | |||
| Firmicutes↑↓ [12,30,38,39,40,41,42,43] | Clostridia↓ [43] | Streptococcaceae↑ [11] | |
| Clostridiaceae↓ [43] | Anaerotruncus↓ [43] | ||
| Ruminococcaceae↓ [11,12,39,43] | Ruminococcus↑↓ [42,43,45,46,47] | ||
| Flavonifractor↑ [38] | |||
| Subdoligranulum↓ [38] | |||
| Faecalibacterium↓ [12,46,48] | |||
| Oscillospira↓ [42] | |||
| Peptostreptococcaceae↓ [43] | |||
| Lachnospiraceae↑↓ [11,12,38,39,42,43] | Lachnospiraceae incertae sedis↑ [11] | ||
| Robinsoniella↑ [39] | |||
| Dorea↑ [39,42] | |||
| Coprococcus↓ [12,38,43,46,47] | |||
| Moryella↓ [43] | |||
| Pseudobutyrivibrio↓ [43] | |||
| Anaerosporobacter↓ [43] | |||
| Roseburia↑↓ [12,39,43] | |||
| Blautia↑↓ [11,12,42,45] | |||
| Peptoniphilaceae | Peptoniphilus↑ [42,43] | ||
| Clostridiales family XI. incertae sedis | Anaerococcus↑ [42] | ||
| Eubacteriaceae | Eubacterium↓ [12,38] | ||
| Oscillospiraceae | Oscillibacter↑↓ [38,39,42] | ||
| Negativicutes | Veillonellaceae↑ [39] | Allisonella↑ [41] | |
| Erysipelotrichia | Erysipelotrichaceae↑ [11] | Holdemania↓ [38] | |
| Bacilli | Lactobacillaceae↑↓ [43,46] | Lactobacillus↑↓ [39,43,46] | |
| Acidaminococcaceae | Acidaminococcus↑ [38] | ||
| Actinobacteria↑↓ [12,38,42] | Actinobacteria | Bifidobacteriaceae↓ [12] | Bifidobacterium↑↓ [12,38] |
| Coriobacteriaceae | Eggerthella↑ [38] |
3.2. Liver and Circulatory Microbiome
4. Gut–Liver Axis—Bidirectional Link
4.1. Definition
4.2. Intestinal Barrier Dysfunction
4.3. Liver and Immune System
5. Gut-Derived Factors (Microbial, Dietary, and Host-Derived)
5.1. Microbiota-Derived Components
5.2. Fructose
5.3. Choline and Its Metabolites
5.4. Short Chain Fatty Acids
5.5. Ethanol and its Metabolites
6. Liver-Derived Factors
6.1. Bile Acids and Their Metabolites
6.2. Immunoglobulin A
7. Conventional Noninvasive Diagnostic and Prognostic Methods
8. Microbiota-Based Biomarkers and Therapeutic Interventions
8.1. Microbiota-Based Diagnostics and Biomarkers
8.2. Microbiota-Based Therapies
9. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- European Association for the Study of the Liver (EASL); European Association for the Study of Diabetes (EASD); European Association for the Study of Obesity (EASO). EASL-EASD-EASO Clinical Practice Guidelines for the Management of Non-Alcoholic Fatty Liver Disease. Obes. Facts 2016, 9, 65–90. [Google Scholar] [CrossRef] [Green Version]
- Bajaj, J.S. Alcohol, liver disease and the gut microbiota. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 235–246. [Google Scholar] [CrossRef]
- Ballestri, S.; Nascimbeni, F.; Romagnoli, D.; Lonardo, A. The independent predictors of non-alcoholic steatohepatitis and its individual histological features.: Insulin resistance, serum uric acid, metabolic syndrome, alanine aminotransferase and serum total cholesterol are a clue to pathogenesis and candidate targets for treatment. Hepatol. Res. 2016, 46, 1074–1087. [Google Scholar] [CrossRef]
- Stepanova, M.; Rafiq, N.; Makhlouf, H.; Agrawal, R.; Kaur, I.; Younoszai, Z.; McCullough, A.; Goodman, Z.; Younossi, Z.M. Predictors of All-Cause Mortality and Liver-Related Mortality in Patients with Non-Alcoholic Fatty Liver Disease (NAFLD). Dig. Dis. Sci. 2013, 58, 3017–3023. [Google Scholar] [CrossRef]
- Vanni, E.; Bugianesi, E.; Kotronen, A.; De Minicis, S.; Yki-Järvinen, H.; Svegliati-Baroni, G. From the metabolic syndrome to NAFLD or vice versa? Dig. Liver Dis. 2010, 42, 320–330. [Google Scholar] [CrossRef] [Green Version]
- Athyros, V.G.; Alexandrides, T.K.; Karagiannis, A.; Karvounis, C.; Katsiki, N.; Kotsis, V.; Kountouras, J.; Liberopoulos, E.; Pitsavos, C.; Polyzos, S.; et al. The use of statins alone, or in combination with pioglitazone and other drugs, for the treatment of non-alcoholic fatty liver disease/non-alcoholic steatohepatitis and related cardiovascular risk. An Expert Panel Statement. Metabolism 2017, 71, 17–32. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Younossi, Z.; Anstee, Q.M.; Marietti, M.; Hardy, T.; Henry, L.; Eslam, M.; George, J.; Bugianesi, E. Global burden of NAFLD and NASH: Trends, predictions, risk factors and prevention. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Betrapally, N.S.; Gillevet, P.M.; Bajaj, J.S. Gut microbiome and liver disease. Transl. Res. 2017, 179, 49–59. [Google Scholar] [CrossRef] [Green Version]
- Carding, S.; Verbeke, K.; Vipond, D.T.; Corfe, B.M.; Owen, L.J. Dysbiosis of the gut microbiota in disease. Microb. Ecol. Health Dis. 2015, 26, 26191. [Google Scholar] [CrossRef] [PubMed]
- Shen, F.; Zheng, R.-D.; Sun, X.-Q.; Ding, W.-J.; Wang, X.-Y.; Fan, J.-G. Gut microbiota dysbiosis in patients with non-alcoholic fatty liver disease. Hepatobiliary Pancreat. Dis. Int. 2017, 16, 375–381. [Google Scholar] [CrossRef]
- Zhu, L.; Baker, S.S.; Gill, C.; Liu, W.; Alkhouri, R.; Baker, R.D.; Gill, S.R. Characterization of gut microbiomes in nonalcoholic steatohepatitis (NASH) patients: A connection between endogenous alcohol and NASH. Hepatology 2013, 57, 601–609. [Google Scholar] [CrossRef]
- Shin, N.-R.; Whon, T.W.; Bae, J.-W. Proteobacteria: Microbial signature of dysbiosis in gut microbiota. Trends Biotechnol. 2015, 33, 496–503. [Google Scholar] [CrossRef]
- Gomez, A.; Petrzelkova, K.J.; Burns, M.B.; Yeoman, C.J.; Amato, K.R.; Vlckova, K.; Modry, D.; Todd, A.; Robinson, C.A.J.; Remis, M.J.; et al. Gut Microbiome of Coexisting BaAka Pygmies and Bantu Reflects Gradients of Traditional Subsistence Patterns. Cell Rep. 2016, 14, 2142–2153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mancabelli, L.; Milani, C.; Lugli, G.A.; Turroni, F.; Ferrario, C.; Van Sinderen, D.; Ventura, M. Meta-analysis of the human gut microbiome from urbanized and pre-agricultural populations. Environ. Microbiol. 2017, 19, 1379–1390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kostovcikova, K.; Coufal, S.; Galanova, N.; Fajstova, A.; Hudcovic, T.; Kostovcik, M.; Prochazkova, P.; Zakostelska, Z.J.; Cermakova, M.; Sediva, B.; et al. Diet Rich in Animal Protein Promotes Pro-inflammatory Macrophage Response and Exacerbates Colitis in Mice. Front. Immunol. 2019, 10, 919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fajstova, A.; Galanova, N.; Coufal, S.; Malkova, J.; Kostovcik, M.; Cermakova, M.; Pelantova, H.; Kuzma, M.; Sediva, B.; Hudcovic, T.; et al. Diet Rich in Simple Sugars Promotes Pro-Inflammatory Response via Gut Microbiota Alteration and TLR4 Signaling. Cells 2020, 9, 2701. [Google Scholar] [CrossRef]
- Hrncirova, L.; Machova, V.; Trckova, E.; Krejsek, J.; Hrncir, T. Food Preservatives Induce Proteobacteria Dysbiosis in Human-Microbiota Associated Nod2-Deficient Mice. Microorganisms 2019, 7, 383. [Google Scholar] [CrossRef] [Green Version]
- Hrncirova, L.; Hudcovic, T.; Sukova, E.; Machova, V.; Trckova, E.; Krejsek, J.; Hrncir, T. Human gut microbes are susceptible to antimicrobial food additives in vitro. Folia Microbiol. 2019, 64, 497–508. [Google Scholar] [CrossRef]
- Chassaing, B.; Van De Wiele, T.; De Bodt, J.; Marzorati, M.; Gewirtz, A.T. Dietary emulsifiers directly alter human microbiota composition and gene expression ex vivo potentiating intestinal inflammation. Gut 2017, 66, 1414–1427. [Google Scholar] [CrossRef]
- Rodriguez-Palacios, A.; Harding, A.; Menghini, P.; Himmelman, C.; Retuerto, M.; Nickerson, K.P.; Lam, M.; Croniger, C.M.; McLean, M.H.; Durum, S.K.; et al. The Artificial Sweetener Splenda Promotes Gut Proteobacteria, Dysbiosis, and Myeloperoxidase Reactivity in Crohn’s Disease–Like Ileitis. Inflamm. Bowel Dis. 2018, 24, 1005–1020. [Google Scholar] [CrossRef]
- Gatea, F.; Sârbu, I.; Vamanu, E. In Vitro Modulatory Effect of Stevioside, as a Partial Sugar Replacer in Sweeteners, on Human Child Microbiota. Microorganisms 2021, 9, 590. [Google Scholar] [CrossRef] [PubMed]
- Suez, J.; Korem, T.; Kuperman, Y.; Harmelin, A.; Kolodkin-Gal, I.; Shapiro, H.; Halpern, Z.; Segal, E.; Elinav, E.; Zeevi, D.; et al. Artificial sweeteners induce glucose intolerance by altering the gut microbiota. Nature 2014, 514, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Catanzaro, J.R.; Strauss, J.D.; Bielecka, A.; Porto, A.F.; Lobo, F.M.; Urban, A.; Schofield, W.B.; Palm, N.W. IgA-deficient humans exhibit gut microbiota dysbiosis despite secretion of compensatory IgM. Sci. Rep. 2019, 9, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Levy, M.; Kolodziejczyk, A.A.; Thaiss, C.A.; Elinav, E. Dysbiosis and the immune system. Nat. Rev. Immunol. 2017, 17, 219–232. [Google Scholar] [CrossRef]
- Chu, H.; Duan, Y.; Yang, L.; Schnabl, B. Small metabolites, possible big changes: A microbiota-centered view of non-alcoholic fatty liver disease. Gut 2018, 68, 359–370. [Google Scholar] [CrossRef] [PubMed]
- Llorente, C.; Schnabl, B. The Gut Microbiota and Liver Disease. Cell. Mol. Gastroenterol. Hepatol. 2015, 1, 275–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Png, C.W.; Lindén, S.K.; Gilshenan, K.S.; Zoetendal, E.G.; McSweeney, C.S.; Sly, L.I.; McGuckin, M.A.; Florin, T.H.J. Mucolytic Bacteria with Increased Prevalence in IBD Mucosa Augment In Vitro Utilization of Mucin by Other Bacteria. Am. J. Gastroenterol. 2010, 105, 2420–2428. [Google Scholar] [CrossRef]
- Aron-Wisnewsky, J.; Vigliotti, C.; Witjes, J.; Le, P.; Holleboom, A.G.; Verheij, J.; Nieuwdorp, M.; Clément, K. Gut microbiota and human NAFLD: Disentangling microbial signatures from metabolic disorders. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 279–297. [Google Scholar] [CrossRef]
- Loomba, R.; Seguritan, V.; Li, W.; Long, T.; Klitgord, N.; Bhatt, A.; Dulai, P.S.; Caussy, C.; Bettencourt, R.; Highlander, S.K.; et al. Gut Microbiome-Based Metagenomic Signature for Non-invasive Detection of Advanced Fibrosis in Human Nonalcoholic Fatty Liver Disease. Cell Metab. 2017, 25, 1054–1062.e5. [Google Scholar] [CrossRef]
- Qin, N.; Yang, F.; Li, A.; Prifti, E.; Chen, Y.; Shao, L.; Guo, J.; Le Chatelier, E.; Yao, J.; Wu, L.; et al. Alterations of the human gut microbiome in liver cirrhosis. Nature 2014, 513, 59–64. [Google Scholar] [CrossRef]
- Le Chatelier, E.; Nielsen, T.; Qin, J.; Prifti, E.; Hildebrand, F.; Falony, G.; Almeida, M.; Arumugam, M.; Batto, J.-M.; Kennedy, S.; et al. Richness of human gut microbiome correlates with metabolic markers. Nature 2013, 500, 541–546. [Google Scholar] [CrossRef]
- Karlsson, F.H.; Tremaroli, V.; Nookaew, I.; Bergström, G.; Behre, C.J.; Fagerberg, B.; Nielsen, J.; Bäckhed, F. Gut metagenome in European women with normal, impaired and diabetic glucose control. Nature 2013, 498, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Sokol, H.; Seksik, P.; Furet, J.P.; Firmesse, O.; Nion-Larmurier, I.; Beaugerie, L.; Cosnes, J.; Corthier, G.; Marteau, P.; Doré, J. Low counts of Faecalibacterium prausnitzii in colitis microbiota. Inflamm. Bowel Dis. 2009, 15, 1183–1189. [Google Scholar] [CrossRef]
- Rajilić–Stojanović, M.; Biagi, E.; Heilig, H.G.; Kajander, K.; Kekkonen, R.A.; Tims, S.; de Vos, W.M. Global and Deep Molecular Analysis of Microbiota Signatures in Fecal Samples From Patients with Irritable Bowel Syndrome. Gastroenterology 2011, 141, 1792–1801. [Google Scholar] [CrossRef]
- Aron-Wisnewsky, J.; Prifti, E.; Belda, E.; Ichou, F.; Kayser, B.D.; Dao, M.C.; Verger, E.O.; Hedjazil, L.; Bouillot, J.-L.; Chevallier, J.-M.; et al. Major microbiota dysbiosis in severe obesity: Fate after bariatric surgery. Gut 2019, 68, 70–82. [Google Scholar] [CrossRef] [PubMed]
- Yang, A.-M.; Inamine, T.; Hochrath, K.; Chen, P.; Wang, L.; Llorente, C.; Bluemel, S.; Hartmann, P.; Xu, J.; Koyama, Y.; et al. Intestinal fungi contribute to development of alcoholic liver disease. J. Clin. Investig. 2017, 127, 2829–2841. [Google Scholar] [CrossRef] [Green Version]
- Hoyles, L.; Fernandez-Real, J.-M.; Federici, M.; Serino, M.; Abbott, J.; Charpentier, J.; Heymes, C.; Luque, J.L.; Anthony, E.; Barton, R.H. Molecular phenomics and metagenomics of hepatic steatosis in non-diabetic obese women. Nat. Med. 2018, 24, 1070–1080. [Google Scholar] [CrossRef]
- Raman, M.; Ahmed, I.; Gillevet, P.M.; Probert, C.S.; Ratcliffe, N.M.; Smith, S.; Greenwood, R.; Sikaroodi, M.; Lam, V.; Crotty, P.; et al. Fecal Microbiome and Volatile Organic Compound Metabolome in Obese Humans with Nonalcoholic Fatty Liver Disease. Clin. Gastroenterol. Hepatol. 2013, 11, 868–875.e3. [Google Scholar] [CrossRef] [PubMed]
- Michail, S.; Lin, M.; Frey, M.R.; Fanter, R.; Paliy, O.; Hilbush, B.; Reo, N.V. Altered gut microbial energy and metabolism in children with non-alcoholic fatty liver disease. FEMS Microbiol. Ecol. 2014, 91, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Wong, V.W.-S.; Tse, C.-H.; Lam, T.T.-Y.; Wong, G.L.-H.; Chim, A.M.-L.; Chu, W.C.-W.; Yeung, D.K.-W.; Law, P.T.-W.; Kwan, H.-S.; Yu, J.; et al. Molecular Characterization of the Fecal Microbiota in Patients with Nonalcoholic Steatohepatitis–A Longitudinal Study. PLoS ONE 2013, 8, e62885. [Google Scholar] [CrossRef] [Green Version]
- Del Chierico, F.; Nobili, V.; Vernocchi, P.; Russo, A.; De Stefanis, C.; Gnani, D.; Furlanello, C.; Zandonà, A.; Paci, P.; Capuani, G.; et al. Gut microbiota profiling of pediatric nonalcoholic fatty liver disease and obese patients unveiled by an integrated meta-omics-based approach. Hepatology 2017, 65, 451–464. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Jiang, X.; Cao, M.; Ge, J.; Bao, Q.; Tang, L.; Chen, Y.; Li, L. Altered Fecal Microbiota Correlates with Liver Biochemistry in Nonobese Patients with Non-alcoholic Fatty Liver Disease. Sci. Rep. 2016, 6, 32002. [Google Scholar] [CrossRef] [PubMed]
- Mouzaki, M.; Comelli, E.M.; Arendt, B.M.; Bonengel, J.; Fung, S.K.; Fischer, S.E.; McGilvray, I.D.; Allard, J.P. Intestinal microbiota in patients with nonalcoholic fatty liver disease. Hepatology 2013, 58, 120–127. [Google Scholar] [CrossRef] [Green Version]
- Boursier, J.; Mueller, O.; Hunault, G.; Oberti, F.; Calès, P.; Diehl, A.M.; Barret, M.; Machado, M.V.; Fizanne, L.; Araujo-Perez, F.; et al. The severity of nonalcoholic fatty liver disease is associated with gut dysbiosis and shift in the metabolic function of the gut microbiota. Hepatology 2016, 63, 764–775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Da Silva, H.E.; Teterina, A.; Comelli, E.M.; Taibi, A.; Arendt, B.M.; Fischer, S.E.; Lou, W.; Allard, J.P. Nonalcoholic fatty liver disease is associated with dysbiosis independent of body mass index and insulin resistance. Sci. Rep. 2018, 8, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alferink, L.J.; Radjabzadeh, D.; Erler, N.S.; Vojinovic, D.; Medina-Gomez, C.; Uitterlinden, A.G.; de Knegt, R.J.; Amin, N.; Ikram, M.A.; Janssen, H.L.; et al. Microbiomics, Metabolomics, Predicted Metagenomics, and Hepatic Steatosis in a Population-Based Study of 1355 Adults. Hepatology 2021, 73, 968–982. [Google Scholar] [CrossRef]
- Chen, Y.; Yang, F.; Lu, H.; Wang, B.; Chen, Y.; Lei, D.; Wang, Y.; Zhu, B.; Li, L. Characterization of fecal microbial communities in patients with liver cirrhosis. Hepatology 2011, 54, 562–572. [Google Scholar] [CrossRef] [PubMed]
- Schierwagen, R.; Alvarez-Silva, C.; Madsen, M.S.A.; Kolbe, C.C.; Meyer, C.; Thomas, D.; Uschner, F.E.; Magdaleno, F.; Jansen, C.; Pohlmann, A.; et al. Circulating microbiome in blood of different circulatory compartments. Gut 2019, 68, 578–580. [Google Scholar] [CrossRef] [Green Version]
- Brandtzaeg, P.; Prydz, H. Direct evidence for an integrated function of J chain and secretory component in epithelial transport of immunoglobulins. Nature 1984, 311, 71–73. [Google Scholar] [CrossRef]
- Mestecky, J.; Russell, M.W.; Elson, C.O. Intestinal IgA: Novel views on its function in the defence of the largest mucosal surface. Gut 1999, 44, 2–5. [Google Scholar] [CrossRef] [Green Version]
- Gautreaux, M.D.; Deitch, E.A.; Berg, R.D. T lymphocytes in host defense against bacterial translocation from the gastrointestinal tract. Infect. Immun. 1994, 62, 2874–2884. [Google Scholar] [CrossRef] [Green Version]
- Johnson, R.J.; Rivard, C.; Lanaspa, M.A.; Otabachian-Smith, S.; Ishimoto, T.; Cicerchi, C.; Cheeke, P.R.; MacIntosh, B.; Hess, T. Fructokinase, Fructans, Intestinal Permeability, and Metabolic Syndrome: An Equine Connection? J. Equine Veter. Sci. 2013, 33, 120–126. [Google Scholar] [CrossRef] [Green Version]
- Spruss, A.; Bergheim, I. Dietary fructose and intestinal barrier: Potential risk factor in the pathogenesis of nonalcoholic fatty liver disease. J. Nutr. Biochem. 2009, 20, 657–662. [Google Scholar] [CrossRef] [PubMed]
- Bergheim, I.; Weber, S.; Vos, M.; Krämer, S.; Volynets, V.; Kaserouni, S.; McClain, C.J.; Bischoff, S.C. Antibiotics protect against fructose-induced hepatic lipid accumulation in mice: Role of endotoxin. J. Hepatol. 2008, 48, 983–992. [Google Scholar] [CrossRef] [PubMed]
- Crispe, I.N. Liver antigen-presenting cells. J. Hepatol. 2011, 54, 357–365. [Google Scholar] [CrossRef] [PubMed]
- Horst, A.K.; Neumann, K.; Diehl, L.; Tiegs, G. Modulation of liver tolerance by conventional and nonconventional antigen-presenting cells and regulatory immune cells. Cell. Mol. Immunol. 2016, 13, 277–292. [Google Scholar] [CrossRef]
- Karimi, M.H.; Geramizadeh, B.; Malek-Hosseini, S.A. Tolerance Induction in Liver. Int. J. Organ Transplant. Med. 2015, 6, 45–54. [Google Scholar]
- Breous, E.; Somanathan, S.; Vandenberghe, L.H.; Wilson, J.M. Hepatic regulatory T cells and Kupffer cells are crucial mediators of systemic T cell tolerance to antigens targeting murine liver. Hepatology 2009, 50, 612–621. [Google Scholar] [CrossRef] [Green Version]
- Carambia, A.; Freund, B.; Schwinge, D.; Heine, M.; Laschtowitz, A.; Huber, S.; Wraith, D.C.; Korn, T.; Schramm, C.; Lohse, A.W.; et al. TGF-β-dependent induction of CD4+CD25+Foxp3+ Tregs by liver sinusoidal endothelial cells. J. Hepatol. 2014, 61, 594–599. [Google Scholar] [CrossRef]
- Crispe, I.N. Immune tolerance in liver disease. Hepatology 2014, 60, 2109–2117. [Google Scholar] [CrossRef] [Green Version]
- Doherty, D.G. Antigen-specific immune tolerance in the liver. Nat. Biomed. Eng. 2019, 3, 763–765. [Google Scholar] [CrossRef]
- Isayama, F.; Hines, I.N.; Kremer, M.; Milton, R.J.; Byrd, C.L.; Perry, A.W.; McKim, S.E.; Parsons, C.; Rippe, R.A.; Wheeler, M.D. LPS signaling enhances hepatic fibrogenesis caused by experimental cholestasis in mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2006, 290, G1318–G1328. [Google Scholar] [CrossRef] [PubMed]
- Gäbele, E.; Mühlbauer, M.; Dorn, C.; Weiss, T.S.; Froh, M.; Schnabl, B.; Wiest, R.; Schölmerich, J.; Obermeier, F.; Hellerbrand, C. Role of TLR9 in hepatic stellate cells and experimental liver fibrosis. Biochem. Biophys. Res. Commun. 2008, 376, 271–276. [Google Scholar] [CrossRef] [PubMed]
- Lebeaupin, C.; Proics, E.; De Bieville, C.H.D.; Rousseau, D.; Bonnafous, S.; Patouraux, S.; Adam, G.; Lavallard, V.J.; Rovere, C.; Le Thuc, O.; et al. ER stress induces NLRP3 inflammasome activation and hepatocyte death. Cell Death Dis. 2015, 6, e1879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghazarian, M.; Revelo, X.S.; Nøhr, M.K.; Luck, H.; Zeng, K.; Lei, H.; Tsai, S.; Schroer, S.A.; Park, Y.J.; Chng, M.H.Y.; et al. Type I interferon responses drive intrahepatic T cells to promote metabolic syndrome. Sci. Immunol. 2017, 2, eaai7616. [Google Scholar] [CrossRef] [Green Version]
- Studer, N.; Desharnais, L.; Beutler, M.; Brugiroux, S.; Terrazos, M.A.; Menin, L.; Schürch, C.M.; McCoy, K.D.; Kuehne, S.A.; Minton, N.P.; et al. Functional Intestinal Bile Acid 7α-Dehydroxylation by Clostridium scindens Associated with Protection from Clostridium difficile Infection in a Gnotobiotic Mouse Model. Front. Cell. Infect. Microbiol. 2016, 6, 191. [Google Scholar] [CrossRef] [Green Version]
- Zhu, W.; Gregory, J.C.; Org, E.; Buffa, J.A.; Gupta, N.; Wang, Z.; Li, L.; Fu, X.; Wu, Y.; Mehrabian, M.; et al. Gut Microbial Metabolite TMAO Enhances Platelet Hyperreactivity and Thrombosis Risk. Cell 2016, 165, 111–124. [Google Scholar] [CrossRef] [Green Version]
- Yajima, M.; Karaki, S.-I.; Tsuruta, T.; Kimura, S.; Nio-Kobayashi, J.; Kuwahara, A.; Yajima, T. Diversity of the intestinal microbiota differently affects non-neuronal and atropine-sensitive ileal contractile responses to short-chain fatty acids in mice. Biomed. Res. 2016, 37, 319–328. [Google Scholar] [CrossRef] [Green Version]
- Johnson, R.J.; Segal, M.S.; Sautin, Y.; Nakagawa, T.; Feig, D.I.; Kang, D.-H.; Gersch, M.S.; Benner, S.; Sánchez-Lozada, L.G. Potential role of sugar (fructose) in the epidemic of hypertension, obesity and the metabolic syndrome, diabetes, kidney disease, and cardiovascular disease. Am. J. Clin. Nutr. 2007, 86, 899–906. [Google Scholar]
- Jensen, T.; Abdelmalek, M.F.; Sullivan, S.; Nadeau, K.J.; Green, M.; Roncal, C.; Nakagawa, T.; Kuwabara, M.; Sato, Y.; Kang, D.-H.; et al. Fructose and sugar: A major mediator of non-alcoholic fatty liver disease. J. Hepatol. 2018, 68, 1063–1075. [Google Scholar] [CrossRef] [Green Version]
- Li, J.-M.; Yu, R.; Zhang, L.-P.; Wen, S.-Y.; Wang, S.-J.; Zhang, X.-Y.; Xu, Q.; Kong, L.-D. Dietary fructose-induced gut dysbiosis promotes mouse hippocampal neuroinflammation: A benefit of short-chain fatty acids. Microbiome 2019, 7, 98. [Google Scholar] [CrossRef] [PubMed]
- Mäenpää, P.H.; Raivio, K.O.; Kekomäki, M.P. Liver Adenine Nuldeotides: Fructose-Induced Depletion and Its Effect on Protein Synthesis. Science 1968, 161, 1253–1254. [Google Scholar] [CrossRef] [PubMed]
- Abdelmalek, M.F.; Lazo, M.; The Fatty Liver Subgroup of the Look AHEAD Research Group; Horska, A.; Bonekamp, S.; Lipkin, E.W.; Balasubramanyam, A.; Bantle, J.P.; Johnson, R.J.; Diehl, A.M.; et al. Higher dietary fructose is associated with impaired hepatic adenosine triphosphate homeostasis in obese individuals with type 2 diabetes. Hepatology 2012, 56, 952–960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bawden, S.; Stephenson, M.; Ciampi, E.; Hunter, K.; Marciani, L.; Macdonald, I.; Aithal, G.; Morris, P.; Gowland, P. Investigating the effects of an oral fructose challenge on hepatic ATP reserves in healthy volunteers: A 31P MRS study. Clin. Nutr. 2016, 35, 645–649. [Google Scholar] [CrossRef] [Green Version]
- Berghe, G.V.D. Fructose: Metabolism and short-term effects on carbohydrate and purine metabolic pathways. Prog. Biochem. Pharmacol. 1986, 21, 1–32. [Google Scholar] [PubMed]
- Le, M.T.; Frye, R.F.; Rivard, C.J.; Cheng, J.; McFann, K.K.; Segal, M.S.; Johnson, R.J.; Johnson, J.A. Effects of high-fructose corn syrup and sucrose on the pharmacokinetics of fructose and acute metabolic and hemodynamic responses in healthy subjects. Metabolism 2012, 61, 641–651. [Google Scholar] [CrossRef] [Green Version]
- Lanaspa, M.A.; Sanchez-Lozada, L.G.; Choi, Y.-J.; Cicerchi, C.; Kanbay, M.; Roncal-Jimenez, C.A.; Ishimoto, T.; Li, N.; Marek, G.; Duranay, M.; et al. Uric Acid Induces Hepatic Steatosis by Generation of Mitochondrial Oxidative Stress: Potential Role in Fructose-Dependent and- Independent Fatty Liver. J. Biol. Chem. 2012, 287, 40732–40744. [Google Scholar] [CrossRef] [Green Version]
- Lim, J.S.; Mietus-Snyder, M.; Valente, A.; Schwarz, J.-M.; Lustig, R.H. The role of fructose in the pathogenesis of NAFLD and the metabolic syndrome. Nat. Rev. Gastroenterol. Hepatol. 2010, 7, 251–264. [Google Scholar] [CrossRef]
- Lanaspa, M.A.; Cicerchi, C.; Garcia, G.; Li, N.; Roncal-Jimenez, C.A.; Rivard, C.J.; Hunter, B.; Andrés-Hernando, A.; Ishimoto, T.; Sánchez-Lozada, L.G.; et al. Counteracting Roles of AMP Deaminase and AMP Kinase in the Development of Fatty Liver. PLoS ONE 2012, 7, e48801. [Google Scholar] [CrossRef] [Green Version]
- Yao, Z.; Vance, D.E. Reduction in VLDL, but not HDL, in plasma of rats deficient in choline. Biochem. Cell Biol. 1990, 68, 552–558. [Google Scholar] [CrossRef]
- Blumberg, H.; Mccollum, E.V.; Albanese, A.A.; Buschke, W. The prevention by choline of liver cirrhosis in rats on high fat, low protein diets. Science 1941, 93, 598–599. [Google Scholar] [CrossRef] [PubMed]
- Sanders, L.M.; Zeisel, S.H. Choline: Dietary Requirements and Role in Brain Development. Nutr. Today 2007, 42, 181–186. [Google Scholar] [CrossRef]
- Shaw, G.M.; Finnell, R.H.; Blom, H.J.; Carmichael, S.L.; Vollset, S.E.; Yang, W.; Ueland, P.M. Choline and Risk of Neural Tube Defects in a Folate-fortified Population. Epidemiology 2009, 20, 714–719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rath, S.; Heidrich, B.; Pieper, D.H.; Vital, M. Uncovering the trimethylamine-producing bacteria of the human gut microbiota. Microbiome 2017, 5, 54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeisel, S.H.; Dacosta, K.A.; Youssef, M.; Hensey, S. Conversion of Dietary Choline to Trimethylamine and Dimethylamine in Rats: Dose-Response Relationship. J. Nutr. 1989, 119, 800–804. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Klipfell, E.; Wu, Y.; Schauer, P.; Smith, J.D.; Allayee, H.; Tang, W.H.W.; DiDonato, J.A.; Lusis, A.J.; Hazen, S.L.; et al. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature 2011, 472, 57–63. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.-M.; Liu, Y.; Zhou, R.-F.; Chen, X.-L.; Wang, C.; Tan, X.-Y.; Wang, L.-J.; Zheng, R.-D.; Zhang, H.-W.; Ling, W.-H.; et al. Associations of gut-flora-dependent metabolite trimethylamine-N-oxide, betaine and choline with non-alcoholic fatty liver disease in adults. Sci. Rep. 2016, 6, 19076. [Google Scholar] [CrossRef]
- Dumas, M.-E.; Barton, R.H.; Mitchell, S.C.; Holmes, E.; McCarthy, M.I.; Scott, J.; Gauguier, D.; Nicholson, J.K.; Toye, A.; Cloarec, O.; et al. Metabolic profiling reveals a contribution of gut microbiota to fatty liver phenotype in insulin-resistant mice. Proc. Natl. Acad. Sci. USA 2006, 103, 12511–12516. [Google Scholar] [CrossRef] [Green Version]
- Gao, X.; Liu, X.; Xu, J.; Xue, C.; Xue, Y.; Wang, Y. Dietary trimethylamine N-oxide exacerbates impaired glucose tolerance in mice fed a high fat diet. J. Biosci. Bioeng. 2014, 118, 476–481. [Google Scholar] [CrossRef]
- Koeth, R.A.; Wang, Z.; Levison, B.S.; Buffa, J.A.; Org, E.; Sheehy, B.T.; Britt, E.B.; Fu, X.; Wu, Y.; Li, L.; et al. Intestinal microbiota metabolism of l-carnitine, a nutrient in red meat, promotes atherosclerosis. Nat. Med. 2013, 19, 576–585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baxter, N.T.; Schmidt, A.W.; Venkataraman, A.; Kim, K.S.; Waldron, C.; Schmidt, T.M. Dynamics of Human Gut Microbiota and Short-Chain Fatty Acids in Response to Dietary Interventions with Three Fermentable Fibers. mBio 2019, 10, e02566-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perry, R.J.; Peng, L.; Barry, N.A.; Cline, G.W.; Zhang, D.; Cardone, R.L.; Petersen, K.F.; Kibbey, R.G.; Goodman, N.A.B.A.L.; Shulman, R.J.P.L.P.G.W.C.R.L.C.K.F.P.R.G.K.G.I. Acetate mediates a microbiome–brain–β-cell axis to promote metabolic syndrome. Nat. Cell Biol. 2016, 534, 213–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vinolo, M.A.R.; Rodrigues, H.G.; Fock, R.A.; Malheiros, G.; Dos Santos, M.F.; Curi, R.; Festuccia, W.T.L.; Crisma, A.R.; Alves, V.S.; Martins, A.R.; et al. Tributyrin attenuates obesity-associated inflammation and insulin resistance in high-fat-fed mice. Am. J. Physiol. Endocrinol. Metab. 2012, 303, E272–E282. [Google Scholar] [CrossRef] [Green Version]
- Weitkunat, K.; Stuhlmann, C.; Postel, A.; Rumberger, S.; Fankhänel, M.; Woting, A.; Petzke, K.J.; Gohlke, S.; Schulz, T.J.; Blaut, M.; et al. Short-chain fatty acids and inulin, but not guar gum, prevent diet-induced obesity and insulin resistance through differential mechanisms in mice. Sci. Rep. 2017, 7, 1–13. [Google Scholar] [CrossRef]
- Park, J.; Kim, M.; Kang, S.G.; Jannasch, A.H.; Cooper, B.; Patterson, J.; Kim, C.H. Short-chain fatty acids induce both effector and regulatory T cells by suppression of histone deacetylases and regulation of the mTOR–S6K pathway. Mucosal Immunol. 2015, 8, 80–93. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.H.; Kang, S.G.; Park, J.H.; Yanagisawa, M.; Kim, C.H. Short-Chain Fatty Acids Activate GPR41 and GPR43 on Intestinal Epithelial Cells to Promote Inflammatory Responses in Mice. Gastroenterology 2013, 145, 396–406.e10. [Google Scholar] [CrossRef]
- Balmer, M.L.; Ma, E.H.; Bantug, G.R.; Grählert, J.; Pfister, S.; Glatter, T.; Jauch, A.; Dimeloe, S.; Slack, E.; Dehio, P.; et al. Memory CD8+ T Cells Require Increased Concentrations of Acetate Induced by Stress for Optimal Function. Immunity 2016, 44, 1312–1324. [Google Scholar] [CrossRef] [Green Version]
- Müller, M.; Hernández, M.A.G.; Goossens, G.H.; Reijnders, D.; Holst, J.J.; Jocken, J.W.E.; Van Eijk, H.; Canfora, E.E.; Blaak, E.E. Circulating but not faecal short-chain fatty acids are related to insulin sensitivity, lipolysis and GLP-1 concentrations in humans. Sci. Rep. 2019, 9, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Watanabe-Suzuki, K.; Seno, H.; Ishii, A.; Kumazawa, T.; Suzuki, O. Ultra-sensitive method for determination of ethanol in whole blood by headspace capillary gas chromatography with cryogenic oven trapping. J. Chromatogr. B Biomed. Sci. Appl. 1999, 727, 89–94. [Google Scholar] [CrossRef]
- Yuan, J.; Chen, C.; Cui, J.; Lu, J.; Yan, C.; Wei, X.; Zhao, X.; Li, N.; Li, S.; Xue, G.; et al. Fatty Liver Disease Caused by High-Alcohol-Producing Klebsiella pneumoniae. Cell Metab. 2019, 30, 675–688.e7. [Google Scholar] [CrossRef]
- Jeon, S.; Carr, R. Alcohol effects on hepatic lipid metabolism. J. Lipid Res. 2020, 61, 470–479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raucy, J.L.; Lasker, J.; Ozaki, K.; Zoleta, V. Regulation of CYP2E1 by Ethanol and Palmitic Acid and CYP4A11 by Clofibrate in Primary Cultures of Human Hepatocytes. Toxicol. Sci. 2004, 79, 233–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, P.; Miyamoto, Y.; Mazagova, M.; Lee, K.-C.; Eckmann, L.; Schnabl, B. Microbiota Protects Mice Against Acute Alcohol-Induced Liver Injury. Alcohol. Clin. Exp. Res. 2015, 39, 2313–2323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boyer, J.L. Bile Formation and Secretion. Compr. Phys. 2013, 3, 1035–1078. [Google Scholar] [CrossRef] [Green Version]
- David, L.A.; Maurice, C.F.; Biddinger, S.B.; Dutton, R.J.; Turnbaugh, P.J.; Carmody, R.N.; Gootenberg, D.B.; Button, J.E.; Wolfe, B.E.; Ling, A.V.; et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature 2014, 505, 559–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Copple, B.L.; Li, T. Pharmacology of bile acid receptors: Evolution of bile acids from simple detergents to complex signaling molecules. Pharmacol. Res. 2016, 104, 9–21. [Google Scholar] [CrossRef] [Green Version]
- Sinal, C.J.; Tohkin, M.; Miyata, M.; Ward, J.M.; Lambert, G.; Gonzalez, F.J. Targeted Disruption of the Nuclear Receptor FXR/BAR Impairs Bile Acid and Lipid Homeostasis. Cell 2000, 102, 731–744. [Google Scholar] [CrossRef] [Green Version]
- Keitel, V.; Donner, M.; Winandy, S.; Kubitz, R.; Häussinger, D. Expression and function of the bile acid receptor TGR5 in Kupffer cells. Biochem. Biophys. Res. Commun. 2008, 372, 78–84. [Google Scholar] [CrossRef]
- Neuschwander-Tetri, B.A.; Loomba, R.; Sanyal, A.J.; Lavine, J.E.; Van Natta, M.L.; Abdelmalek, M.F.; Chalasani, N.; Dasarathy, S.; Diehl, A.M.; Hameed, B.; et al. Farnesoid X nuclear receptor ligand obeticholic acid for non-cirrhotic, non-alcoholic steatohepatitis (FLINT): A multicentre, randomised, placebo-controlled trial. Lancet 2015, 385, 956–965. [Google Scholar] [CrossRef] [Green Version]
- Moro-Sibilot, L.; Blanc, P.; Taillardet, M.; Bardel, E.; Couillault, C.; Boschetti, G.; Traverse-Glehen, A.; Defrance, T.; Kaiserlian, D.; Dubois, B. Mouse and Human Liver Contain Immunoglobulin A–Secreting Cells Originating From Peyer’s Patches and Directed Against Intestinal Antigens. Gastroenterology 2016, 151, 311–323. [Google Scholar] [CrossRef] [Green Version]
- Brown, W.R.; Kloppel, T.M. The role of the liver in Translocation of IgA into the Gastrointestinal Tract. Immunol. Investig. 1989, 18, 269–285. [Google Scholar] [CrossRef] [PubMed]
- Rogier, E.W.; Frantz, A.L.; Bruno, M.E.C.; Wedlund, L.; Cohen, D.A.; Stromberg, A.J.; Kaetzel, C.S. Secretory antibodies in breast milk promote long-term intestinal homeostasis by regulating the gut microbiota and host gene expression. Proc. Natl. Acad. Sci. USA 2014, 111, 3074–3079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Juo, Y.-Y.; Livingston, E.H. Testing for Nonalcoholic Fatty Liver Disease. JAMA 2019, 322, 1836. [Google Scholar] [CrossRef] [PubMed]
- Rinella, M.E. Nonalcoholic Fatty Liver Disease. JAMA 2015, 313, 2263–2273. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.; You, H.J.; Bajaj, J.S.; Joo, S.K.; Yu, J.; Park, S.; Kang, H.; Park, J.H.; Kim, J.H.; Lee, D.H.; et al. Distinct signatures of gut microbiome and metabolites associated with significant fibrosis in non-obese NAFLD. Nat. Commun. 2020, 11, 4982. [Google Scholar] [CrossRef] [PubMed]
- Caussy, C.; Hsu, C.; Schork, N.; Schnabl, B.; Brenner, D.A.; Sirlin, C.B.; Chen, C.-H.; Loomba, R.; Genetics of NAFLD in Twins Consortium; Lo, M.-T.; et al. Link between gut-microbiome derived metabolite and shared gene-effects with hepatic steatosis and fibrosis in NAFLD. Hepatology 2018, 68, 918–932. [Google Scholar] [CrossRef] [Green Version]
- Bajaj, J.S.; Salzman, N.H.; Lee, H.; Osman, M.; Siddiqui, M.S.; Fuchs, M.; Puri, P.; Sikaroodi, M.; Gillevet, P.M.; Acharya, C.; et al. Fecal Microbial Transplant Capsules Are Safe in Hepatic Encephalopathy: A Phase 1, Randomized, Placebo-Controlled Trial. Hepatology 2019, 70, 1690–1703. [Google Scholar] [CrossRef]
- Philips, C.A.; Pande, A.; Shasthry, S.M.; Jamwal, K.D.; Khillan, V.; Chandel, S.S.; Kumar, G.; Sharma, M.K.; Maiwall, R.; Jindal, A.; et al. Healthy Donor Fecal Microbiota Transplantation in Steroid-Ineligible Severe Alcoholic Hepatitis: A Pilot Study. Clin. Gastroenterol. Hepatol. 2017, 15, 600–602. [Google Scholar] [CrossRef]
- Degnan, F.H. Clinical studies involving probiotics. Gut Microbes 2012, 3, 485–489. [Google Scholar] [CrossRef] [Green Version]
- Gibson, G.R.; Roberfroid, M.B. Dietary Modulation of the Human Colonic Microbiota: Introducing the Concept of Prebiotics. J. Nutr. 1995, 125, 1401–1412. [Google Scholar] [CrossRef]
- Vamanu, E. Complementary Functional Strategy for Modulation of Human Gut Microbiota. Curr. Pharm. Des. 2019, 24, 4144–4149. [Google Scholar] [CrossRef]
- Gibson, G.R.; Hutkins, R.; Sanders, M.E.; Prescott, S.L.; Reimer, R.A.; Salminen, S.J.; Scott, K.; Stanton, C.; Swanson, K.S.; Cani, P.D.; et al. Expert consensus document: The International Scientific Association for Probiotics and Prebiotics (ISAPP) consensus statement on the definition and scope of prebiotics. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 491–502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharpton, S.R.; Maraj, B.; Harding-Theobald, E.; Vittinghoff, E.; Terrault, N.A. Gut microbiome–targeted therapies in nonalcoholic fatty liver disease: A systematic review, meta-analysis, and meta-regression. Am. J. Clin. Nutr. 2019, 110, 139–149. [Google Scholar] [CrossRef] [PubMed]
- Duan, Y.; Llorente, C.; Lang, S.; Brandl, K.; Chu, H.; Jiang, L.; White, R.C.; Clarke, T.H.; Nguyen, K.; Torralba, M.; et al. Bacteriophage targeting of gut bacterium attenuates alcoholic liver disease. Nature 2019, 575, 505–511. [Google Scholar] [CrossRef]
- Wang, Z.; Roberts, A.B.; Buffa, J.A.; Levison, B.S.; Zhu, W.; Org, E.; Gu, X.; Huang, Y.; Zamanian-Daryoush, M.; Culley, M.K.; et al. Non-lethal Inhibition of Gut Microbial Trimethylamine Production for the Treatment of Atherosclerosis. Cell 2015, 163, 1585–1595. [Google Scholar] [CrossRef] [Green Version]
- Cipriani, S.; Mencarelli, A.; Palladino, G.; Fiorucci, S. FXR activation reverses insulin resistance and lipid abnormalities and protects against liver steatosis in Zucker (fa/fa) obese rats. J. Lipid Res. 2010, 51, 771–784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fickert, P.; Fuchsbichler, A.; Moustafa, T.; Wagner, M.; Zollner, G.; Halilbasic, E.; Stöger, U.; Arrese, M.; Pizarro, M.; Solís, N.; et al. Farnesoid X Receptor Critically Determines the Fibrotic Response in Mice but Is Expressed to a Low Extent in Human Hepatic Stellate Cells and Periductal Myofibroblasts. Am. J. Pathol. 2009, 175, 2392–2405. [Google Scholar] [CrossRef] [Green Version]
- Verbeke, L.; Farre, R.; Laleman, W.; Trebicka, J.; Komuta, M.; Roskams, T.; Klein, S.; Elst, I.V.; Windmolders, P.; Vanuytsel, T.; et al. Obeticholic acid, a farnesoid X receptor agonist, improves portal hypertension by two distinct pathways in cirrhotic rats. Hepatology 2014, 59, 2286–2298. [Google Scholar] [CrossRef] [PubMed]
- Harrison, S.A.; Rinella, M.E.; Abdelmalek, M.F.; Trotter, J.F.; Paredes, A.H.; Arnold, H.L.; Kugelmas, M.; Bashir, M.R.; Jaros, M.J.; Ling, L.; et al. NGM282 for treatment of non-alcoholic steatohepatitis: A multicentre, randomised, double-blind, placebo-controlled, phase 2 trial. Lancet 2018, 391, 1174–1185. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hrncir, T.; Hrncirova, L.; Kverka, M.; Hromadka, R.; Machova, V.; Trckova, E.; Kostovcikova, K.; Kralickova, P.; Krejsek, J.; Tlaskalova-Hogenova, H. Gut Microbiota and NAFLD: Pathogenetic Mechanisms, Microbiota Signatures, and Therapeutic Interventions. Microorganisms 2021, 9, 957. https://doi.org/10.3390/microorganisms9050957
Hrncir T, Hrncirova L, Kverka M, Hromadka R, Machova V, Trckova E, Kostovcikova K, Kralickova P, Krejsek J, Tlaskalova-Hogenova H. Gut Microbiota and NAFLD: Pathogenetic Mechanisms, Microbiota Signatures, and Therapeutic Interventions. Microorganisms. 2021; 9(5):957. https://doi.org/10.3390/microorganisms9050957
Chicago/Turabian StyleHrncir, Tomas, Lucia Hrncirova, Miloslav Kverka, Robert Hromadka, Vladimira Machova, Eva Trckova, Klara Kostovcikova, Pavlina Kralickova, Jan Krejsek, and Helena Tlaskalova-Hogenova. 2021. "Gut Microbiota and NAFLD: Pathogenetic Mechanisms, Microbiota Signatures, and Therapeutic Interventions" Microorganisms 9, no. 5: 957. https://doi.org/10.3390/microorganisms9050957