
Gut Microbiome of Indonesian Adults Associated with Obesity and Type 2 Diabetes: A Cross-Sectional Study in an Asian City, Yogyakarta

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethics Declaration
2.2. Study Design
2.3. Dietary Information
2.4. Fecal Sample Collection and Transportation Process
2.5. 16S rRNA Gene Amplicon Sequencing and Sequence Data Process
2.6. Statistical Analysis
2.7. Alpha-Diversity Analysis
2.8. Beta Diversity Analysis
2.9. Linear Discriminant Analysis Effect Size (LEfSe)
2.10. Nuclear Magnetic Resonance (NMR) Metabolomics
2.11. Fecal Bile Acid Measurement
3. Results
3.1. Physical Characteristics of Indonesian Subjects and Their Trends in Macronutrient Consumption
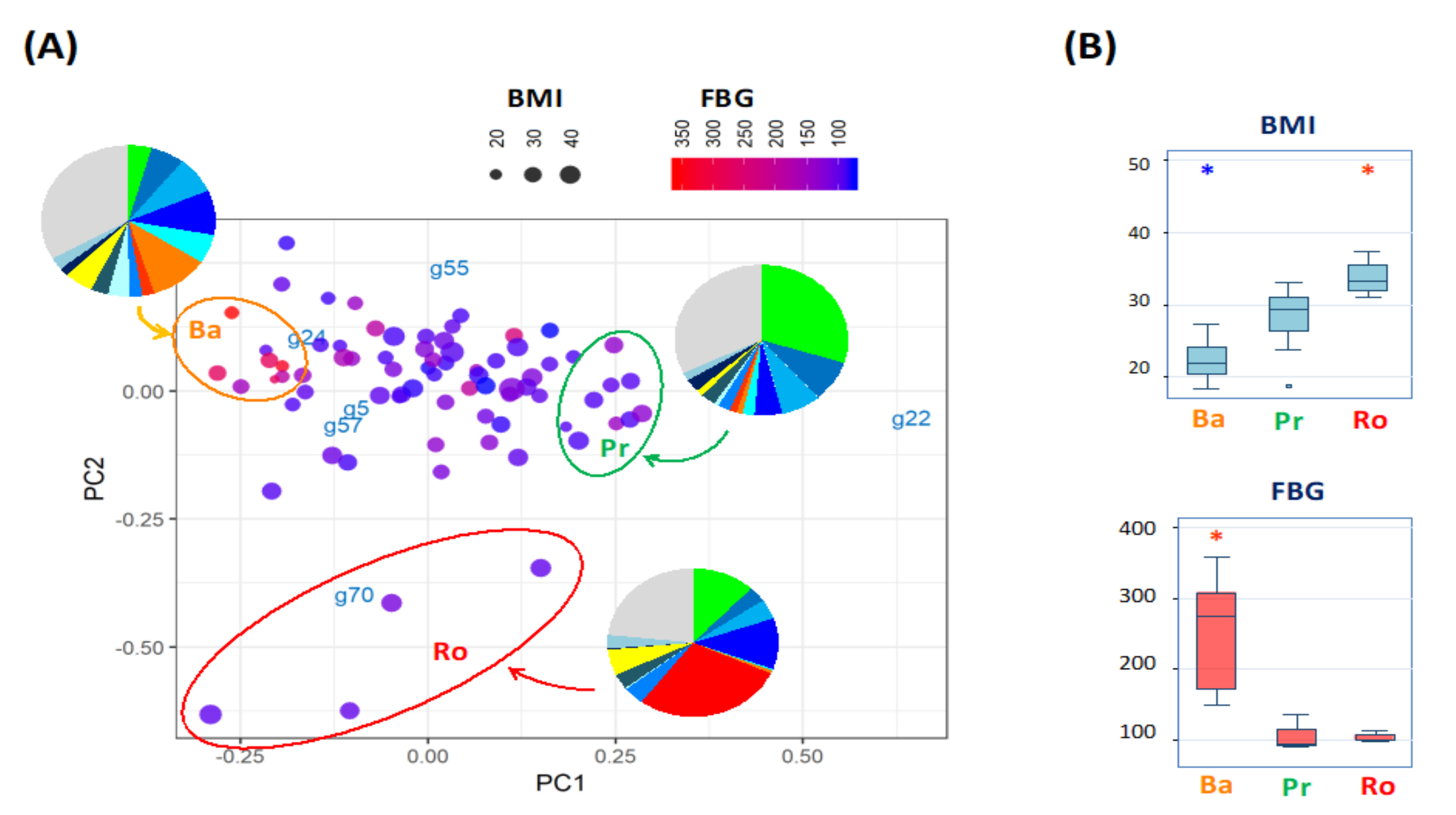
3.2. Gut Microbiome Variance of Indonesian Subjects Is Driven by Three Bacterial Genera
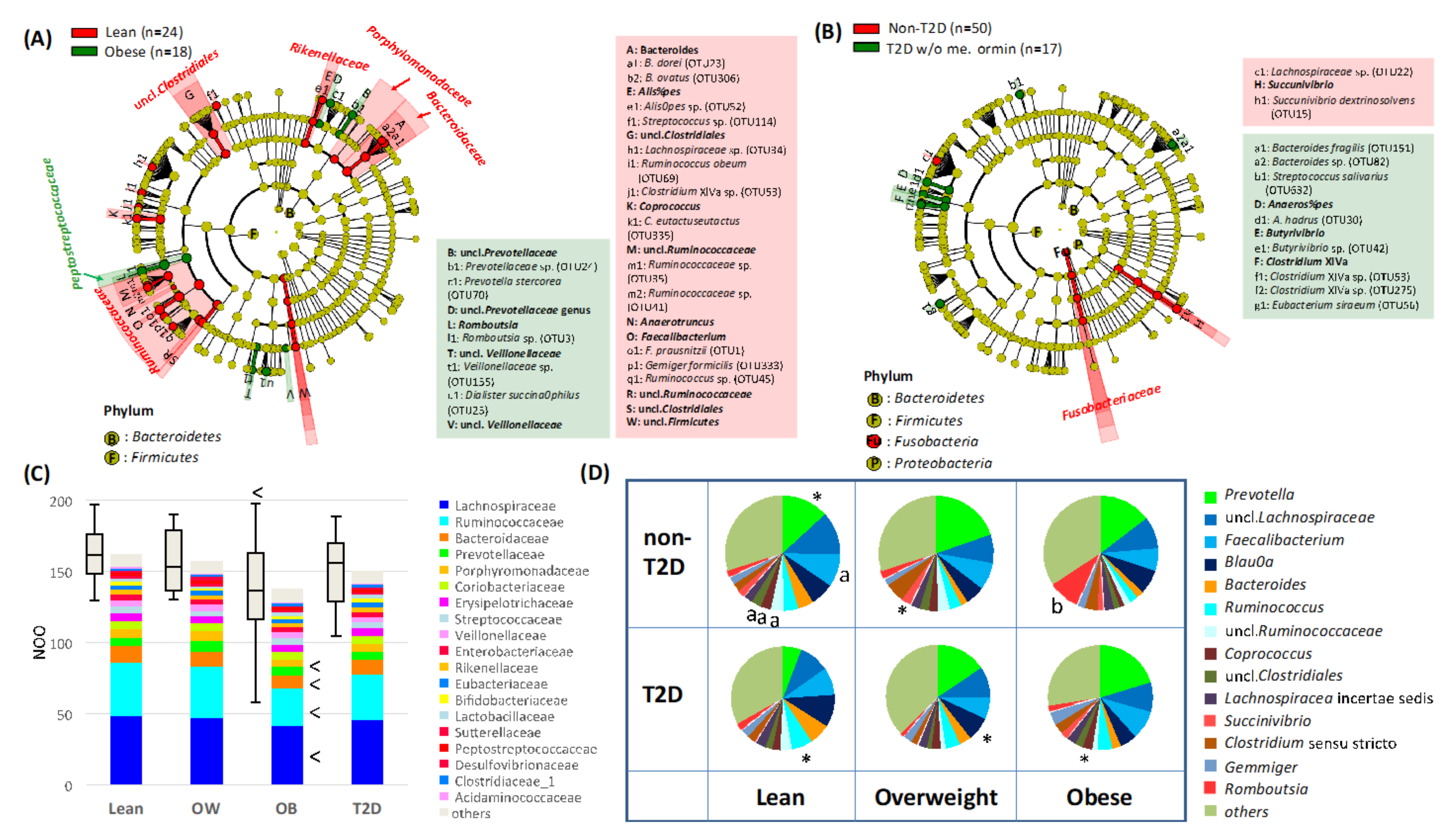
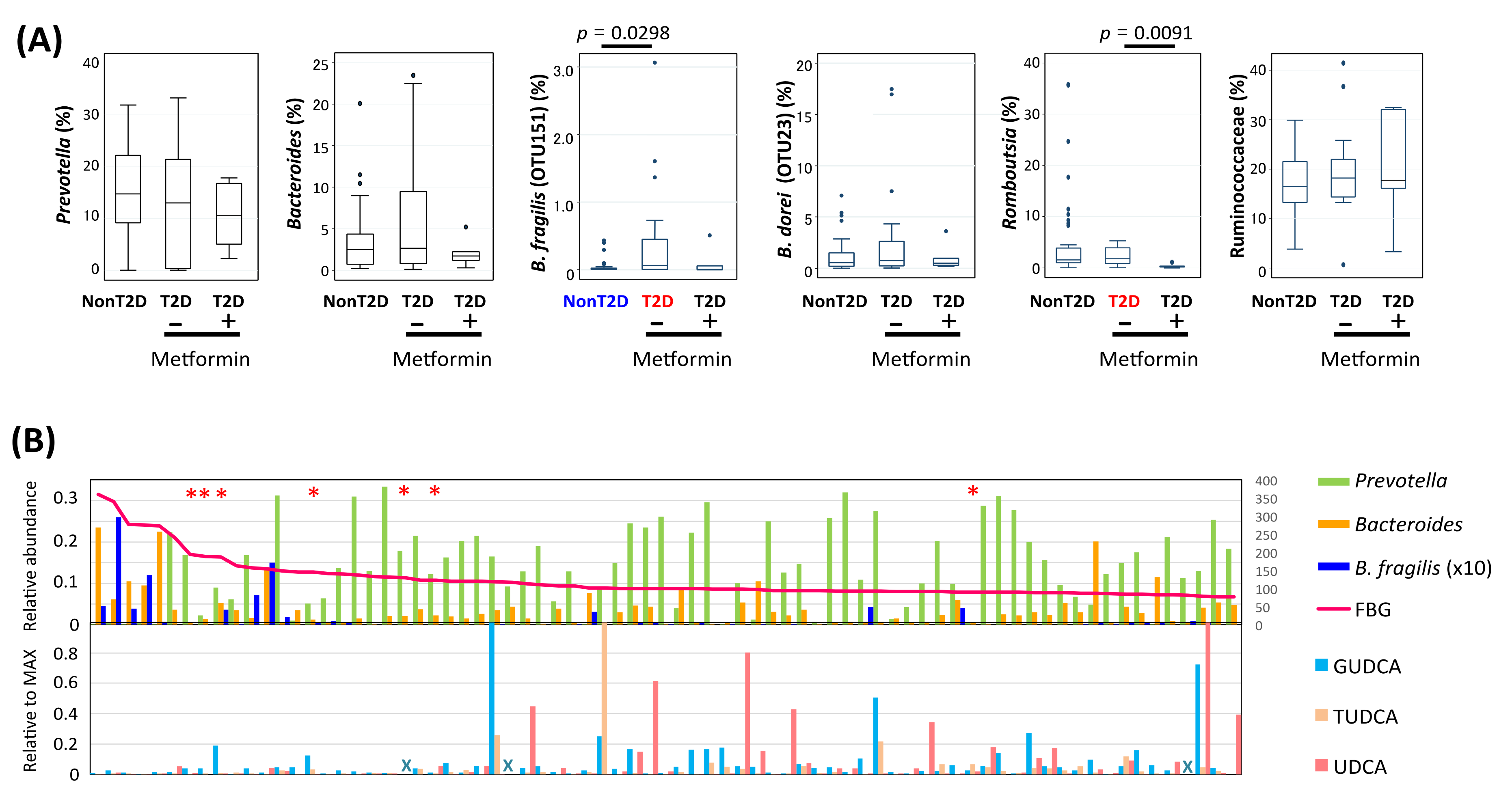
3.3. Gut Microbiome Features of Obese or T2D Indonesian Subjects
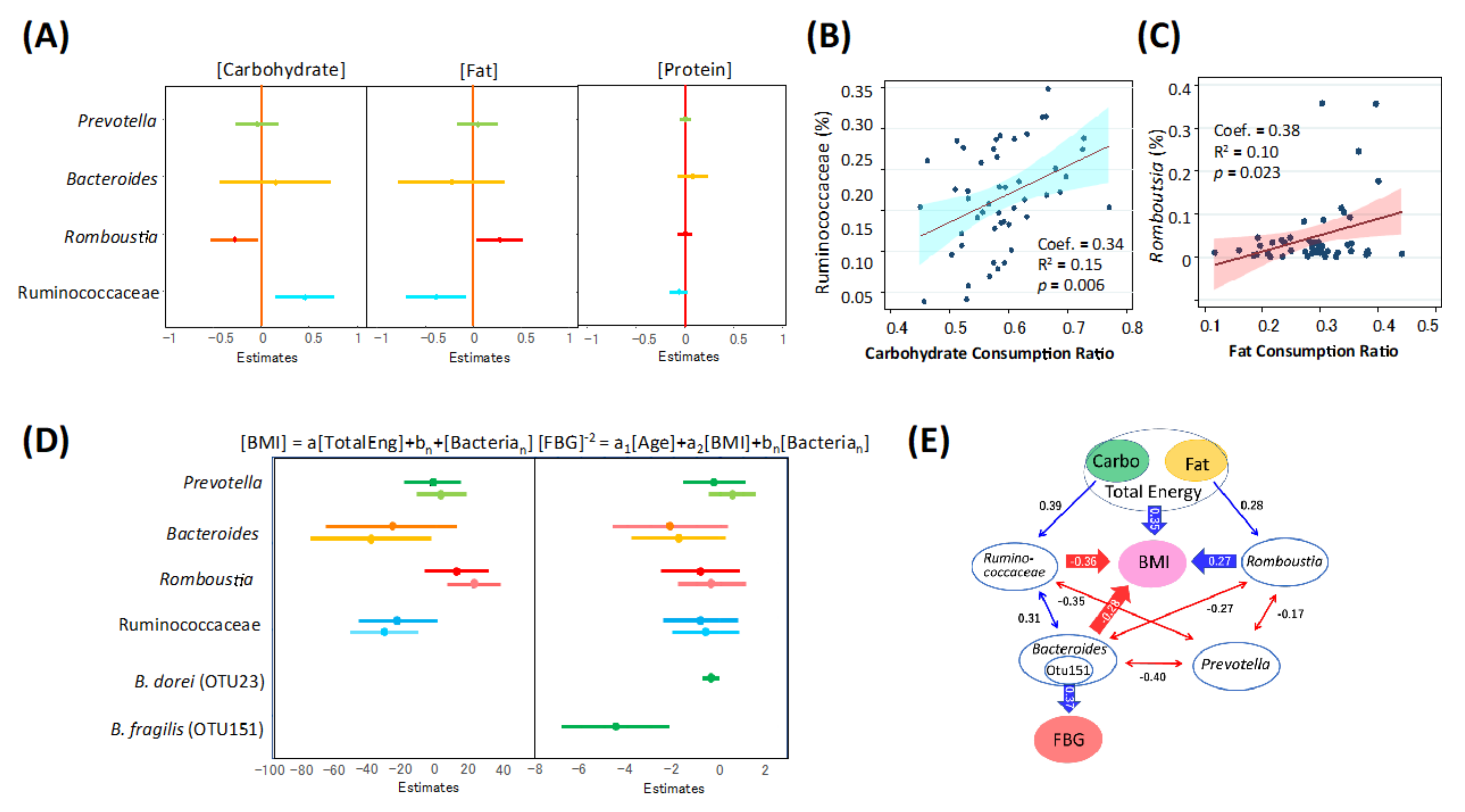
3.4. Association of Gut Microbiome of Indonesian Subjects with Diets, Obesity, and T2D
3.5. Potential Microbiome Markers for Fat-Driven Obesity in Indonesian Subjects
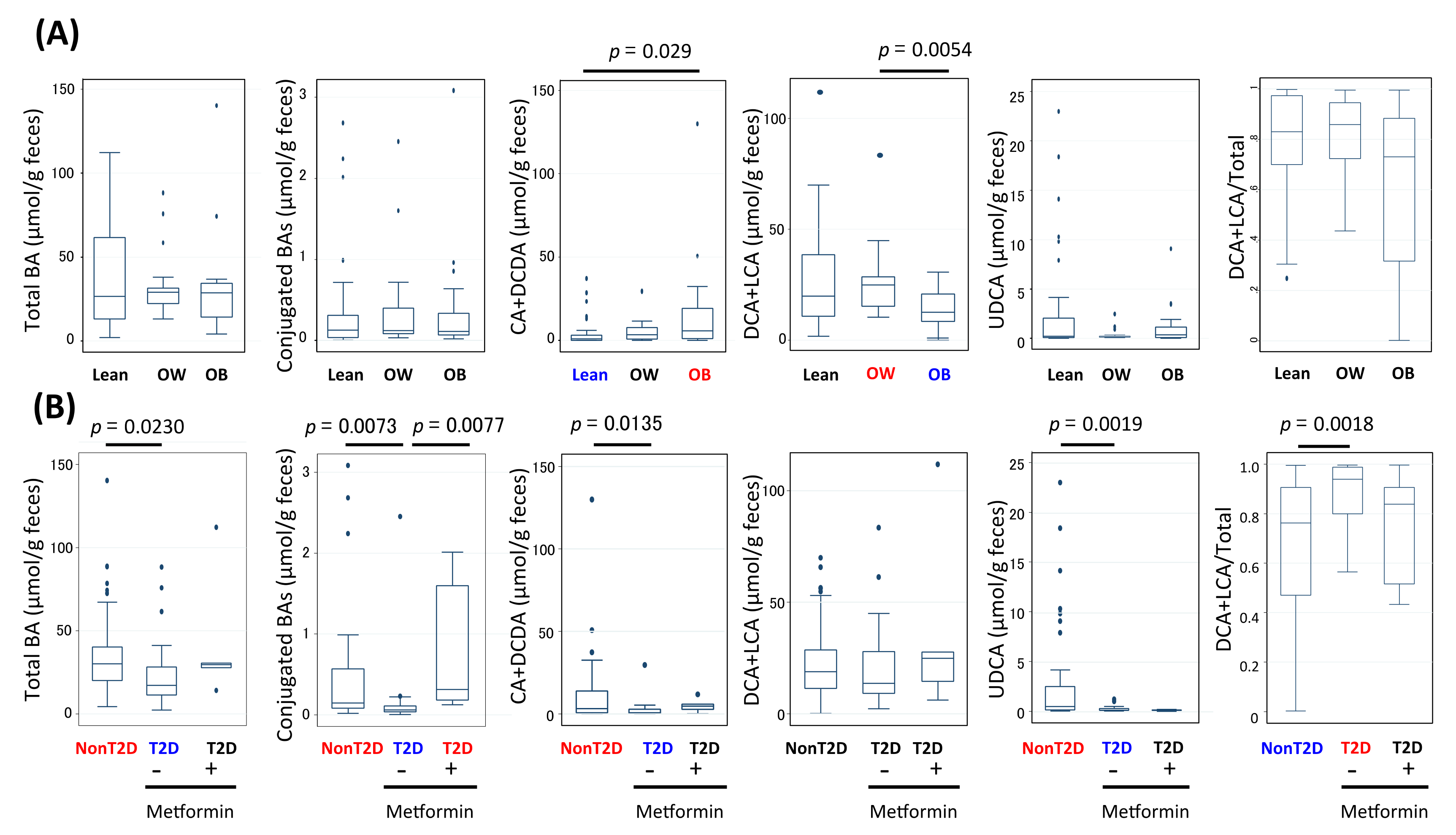
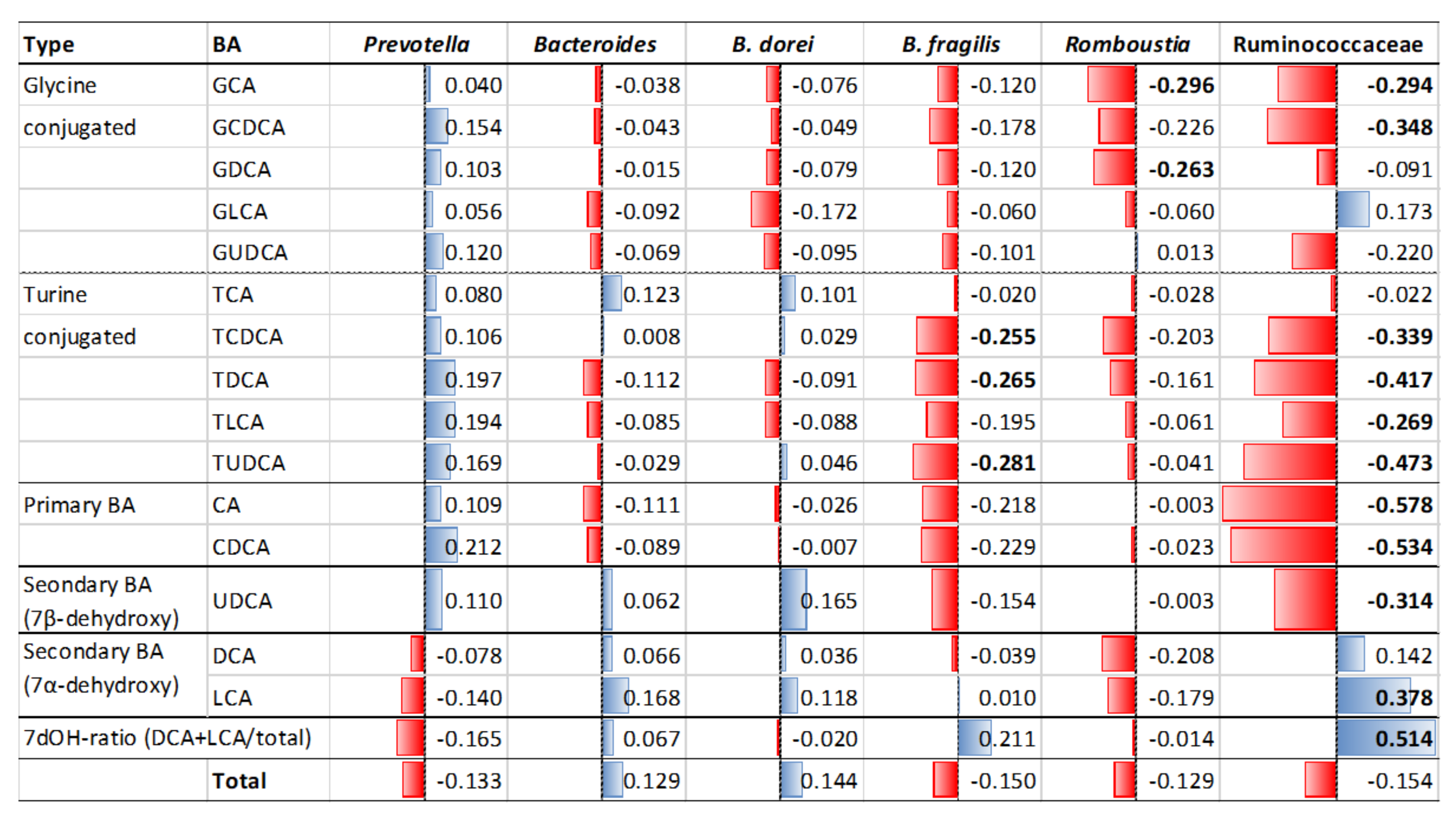
3.6. Alteration of Bile Acid Metabolism in Microbiota of Obese and T2D Indonesian Subjects
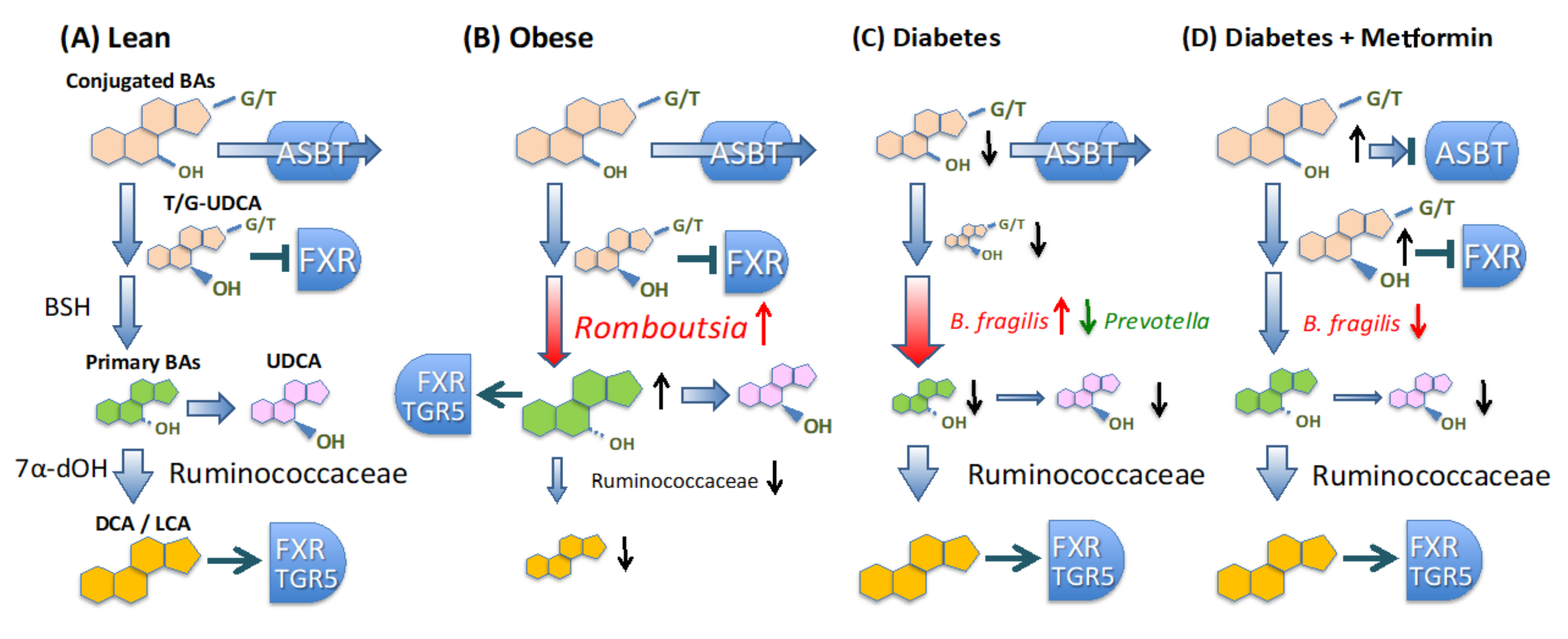
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nakayama, J.; Watanabe, K.; Jiang, J.; Matsuda, K.; Chao, S.H.; Haryono, P.; La-ongkham, O.; Sarwoko, M.A.; Sujaya, N.; Zhao, L.; et al. Diversity in gut bacterial community of school-age children in Asia. Sci. Rep. 2015, 5, 8397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakayama, J.; Yamamoto, A.; Palermo-Conde, L.A.; Higashi, K.; Sonomoto, K.; Tan, J.; Lee, Y.K. Impact of westernized diet on gut microbiota in children on Leyte island. Front. Microbiol. 2017, 8, 197. [Google Scholar] [CrossRef] [PubMed]
- Kisuse, J.; La-ongkham, O.; Nakphaichit, M.; Therdtatha, P.; Momoda, R.; Tanaka, M.; Fukuda, S.; Popluechai, S.; Kespechara, K.; Sonomoto, K.; et al. Urban diets linked to gut microbiome and metabolome alterations in children: A comparative cross-sectional study in Thailand. Front. Microbiol. 2018, 9, 1345. [Google Scholar] [CrossRef] [PubMed]
- Burcelin, R. Gut microbiota and immune crosstalk in metabolic disease. Mol. Metab. 2016, 5, 771–781. [Google Scholar] [CrossRef]
- Gonçalves, P.; Araújo, J.R.; Di Santo, J.P. A cross-talk between microbiota-derived short-chain fatty acids and the host mucosal immune system regulates intestinal homeostasis and inflammatory bowel disease. Inflam. Bowel Dis. 2018, 24, 558–572. [Google Scholar] [CrossRef] [Green Version]
- Le Chatelier, E.L.; Nielsen, T.; Qin, J.; Prifti, E.; Hildebrand, F.; Falony, G.; Almeida, M.; Arumugam, M.; Batto, J.M.; Kennedy, S.; et al. Richness of human gut microbiome correlates with metabolic markers. Nature 2013, 500, 541–546. [Google Scholar] [CrossRef]
- Graessler, J.; Qin, Y.; Zhong, H.; Zhang, J.; Licinio, J.; Wong, M.L.; Xu, A.; Chavakis, T.; Bornstein, A.B.; Ehrhart-Bornstein, M.; et al. Metagenomic sequencing of the human gut microbiome before and after bariatric surgery in obese patients with type 2 diabetes: Correlation with inflammatory and metabolic parameters. Pharm. J 2013, 13, 514–522. [Google Scholar] [CrossRef] [Green Version]
- Qin, J.; Li, Y.; Cai, Z.; Li, S.; Zhu, J.; Zhang, F.; Liang, S.; Zhang, W.; Guan, Y.; Shen, D.; et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature 2012, 490, 55–60. [Google Scholar] [CrossRef]
- Forslund, K.; Hildebrand, F.; Nielsen, T.; Falony, G.; Le Chatelier, E.L.; Sunagawa, S.; Prifti, E.; Vieira-Silva, S.; Gudmundottir, V.; Pedersen, H.K.; et al. Disentangling type 2 diabetes and metformin treatment signatures in the human gut microbiota. Nature 2015, 528, 262–266. [Google Scholar] [CrossRef]
- Boulangé, C.L.; Neves, A.L.; Chilloux, J.; Nicholson, J.K.; Dumas, M.E. Impact of the gut microbiota on inflammation, obesity, and metabolic disease. Genome Med. 2016, 8, 42. [Google Scholar] [CrossRef] [Green Version]
- Ríos-Covián, D.; Ruas-Madiedo, P.; Margolles, A.; Gueimonde, M.; de los Reyes-Gavilán, C.G.; Salazar, N. Intestinal short chain fatty acids and their link with diet and human health. Front. Microbiol. 2016, 7, 185. [Google Scholar] [CrossRef] [Green Version]
- Byrne, C.S.; Chambers, E.S.; Morrison, D.J.; Frost, G. The role of short chain fatty acids in appetite regulation and energy homeostasis. Int. J. Obes. 2015, 39, 1331–1338. [Google Scholar] [CrossRef] [Green Version]
- Fernández-Veledo, S.; Vendrell, J. Gut microbiota-derived succinate: Friend or foe in human metabolic diseases? Rev. Endocr. Metab. Disord. 2019, 20, 439–447. [Google Scholar] [CrossRef] [Green Version]
- Makishima, M.; Okamoto, A.Y.; Repa, J.J.; Tu, H.; Learned, R.M.; Luk, A.; Hull, M.V.; Lustig, K.D.; Mangelsdorf, D.J.; Shan, B. Identification of a nuclear receptor for bile acids. Science 1999, 284, 1362–1365. [Google Scholar] [CrossRef]
- Kawamata, Y.; Fujii, R.; Hosoya, M.; Harada, M.; Yoshida, H.; Miwa, M.; Fukusumi, S.; Habata, Y.; Itoh, T.; Shintani, Y.; et al. A G protein-coupled receptor responsive to bile acids. J. Biol. Chem. 2003, 278, 9435–9440. [Google Scholar] [CrossRef] [Green Version]
- Ferrell, J.M.; Chiang, J.Y.L. Understanding bile acid signaling in diabetes: From pathophysiology to therapeutic targets. Diabetes Metab. J. 2019, 43, 257–272. [Google Scholar] [CrossRef]
- Begley, M.; Gahan, C.G.M.; Hill, C. The interaction between bacteria and bile. FEMS Microbiol. Rev. 2005, 29, 625–651. [Google Scholar] [CrossRef] [Green Version]
- Ramachandran, A.; Chamukuttan, S.; Shetty, S.A.; Arun, N.; Susairaj, P. Obesity in Asia—is it different from rest of the world. Diabetes Metab. Res. Rev. 2012, 28 (Suppl. 2), 47–51. [Google Scholar] [CrossRef]
- Kahn, S.E.; Hull, R.L.; Utzschneider, K.M. Mechanisms linking obesity to insulin resistance and type 2 diabetes. Nature 2006, 444, 840–846. [Google Scholar] [CrossRef]
- Fabbrini, E.; Yoshino, J.; Yoshino, M.; Magkos, F.; Tiemann Luecking, C.T.; Samovski, D.; Fraterrigo, G.; Okunade, A.L.; Patterson, B.W.; Klein, S. Metabolically normal obese people are protected from adverse effects following weight gain. J. Clin. Invest. 2015, 125, 787–795. [Google Scholar] [CrossRef] [Green Version]
- Meigs, J.B.; Wilson, P.W.F.; Fox, C.S.; Vasan, R.S.; Nathan, D.M.; Sullivan, L.M.; D’Agostino, R.B. Body mass index, metabolic syndrome, and risk of type 2 diabetes or cardiovascular disease. J. Clin. Endocrinol. Metab. 2006, 91, 2906–2912. [Google Scholar] [CrossRef]
- Lee, J.W.R.; Brancati, F.L.; Yeh, H.C. Trends in the prevalence of type 2 diabetes in Asians versus whites: Results from the United States National Health Interview Survey, 1997–2008. Diabetes Care 2011, 34, 353–357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, J.C.N.; Malik, V.; Jia, W.; Kadowaki, T.; Yajnik, C.S.; Yoon, K.H.; Hu, F.B. Diabetes in Asia: Epidemiology, risk factors, and pathophysiology. JAMA 2009, 301, 2129–2140. [Google Scholar] [CrossRef] [PubMed]
- Astrup, A.; Finer, N. Redefining type 2 diabetes: “diabesity” or “obesity dependent diabetes mellitus”? Obes. Rev. 2000, 1, 57–59. [Google Scholar] [CrossRef] [PubMed]
- Rhee, E.J. Diabetes in Asians. Endocrinol. Metab. 2015, 30, 263–269. [Google Scholar] [CrossRef] [Green Version]
- Hussain, A. Diabetes in Asia: Special challenges and solutions. J. Diabetol. 2018, 9, 69–71. [Google Scholar] [CrossRef]
- Pan, A.; Malik, V.S.; Hu, F.B. Exporting diabetes mellitus to Asia: The impact of Western-style fast food. Circulation 2012, 126, 163–165. [Google Scholar] [CrossRef] [Green Version]
- De Filippo, C.; Cavalieri, D.; Paola, M.D.; Ramazzotti, M.; Poullet, J.B.; Massart, S.; Collini, S.; Pieraccini, G.; Lionetti, P. Impact of diet in shaping gut microbiota revealed by a comparative study in children from Europe and rural Africa. Proc. Natl. Acad. Sci. USA 2010, 107, 14691–14696. [Google Scholar] [CrossRef] [Green Version]
- Ruengsomwong, S.; La-ongkham, O.; Jiang, J.; Wannissorn, B.; Nakayama, J.; Nitisinprasert, S. Microbial community of healthy Thai vegetarians and non-vegetarians, their core gut microbiota, and pathogen risk. J. Microbiol. Biotechnol. 2016, 26, 1723–1735. [Google Scholar] [CrossRef] [Green Version]
- WHO. World Health Organization-Diabetes Country Profiles; WHO: Geneva, Switzerland, 2016. [Google Scholar]
- International Diabetes Federation. IDF Diabetes Atlas, 9th ed.; International Diabetes Federation: Brussels, Belgium, 2019. [Google Scholar]
- Oddo, V.M.; Maehara, M.; Rah, J.H. Overweight in Indonesia: And observational study of trends and risk factors among adults and children. BMJ Open 2019, 9, e031198. [Google Scholar] [CrossRef]
- Huriyati, E.; Kandarina, B.J.I.; Faza, F. Association of eating pattern and nutritional status dyslipidemia among adults in Yogyakarta-Indonesia. KnE Life Sci. 2019, 4, 86–96. [Google Scholar] [CrossRef]
- Sekiyama, M.; Roosita, K.; Ohtsuka, R. Snack foods consumption contributes to poor nutrition of rural children in West Java. Asia Pac. J. Clin. Nutr. 2012, 21, 558–567. [Google Scholar]
- Zhao, H.; Chen, J.; Li, X.; Sun, Q.; Qin, P.; Wang, Q. Compositional and functional features of the female premenopausal and postmenopausal gut microbiota. FEBS Lett. 2019, 593, 2655–2664. [Google Scholar] [CrossRef]
- WHO. Obesity: Preventing and Managing the Global Epidemic; Report on a WHO Consultation on Obesity; WHO: Geneva, Switzerland, 2000; Volume 894. [Google Scholar]
- Seino, Y.; Nanjo, K.; Tajima, N.; Kadowaki, T.; Kashiwagi, A.; Araki, E.; Ito, C.; Inagaki, N.; Iwamoto, Y.; Kasuga, M.; et al. Report of the Committee on the classification and diagnostic criteria of diabetes mellitus. The Committee of the Japan Diabetes Society on the diagnostic criteria of diabetes mellitus. Diabetol. Int. 2010, 1, 2–20. [Google Scholar] [CrossRef] [Green Version]
- Matsuki, T.; Watanabe, K.; Fujimoto, J.; Takada, T.; Tanaka, R. Use of 16s rRNA gene-targeted group-specific primers for real-time PCR analysis of predominant bacteria in human feces. Appl. Environ. Microbiol. 2004, 70, 7220–7228. [Google Scholar] [CrossRef] [Green Version]
- Herlemann, D.P.; Labrenz, M.; Jürgens, K.; Bertilsson, S.; Waniek, J.J.; Andersson, A.F. Transitions in bacterial communities along the 2000 km salinity gradient of the Baltic Sea. ISME J. 2011, 5, 1571–1579. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.C. Uparse: Highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 2013, 10, 996–998. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. SINTAX: A simple non-Bayesian taxonomy classifier for 16S and ITS sequences. bioRxiv 2016. [Google Scholar] [CrossRef] [Green Version]
- Kuczynski, J.; Stombaugh, J.; Walters, W.A.; González, A.; Caporaso, G.; Knight, R. Using QIIME to analyze 16S rRNA gene sequences from Microbial Communities. Curr. Protoc. Bioinform. 2011, 10, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Shannon, C.E. A mathematical theory of communication. Bell. Syst. Tech. J. 1948, 27, 623–656. [Google Scholar] [CrossRef]
- Faith, D.P.; Baker, A.M. Phylogenetic diversity (PD) and biodiversity conservation: Some bioinformatics challenges. Evol. Bioinform. 2007, 2, 121–128. [Google Scholar] [CrossRef]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, Y.; Ma, C.; Liu, C.; Wang, Z.; Yang, J.; Liu, X.; Shen, Z.; Wu, R. NMR-based fecal metabolomics fingerprinting as predictors of earlier diagnosis inpatients with colorectal cancer. Oncotarget 2016, 7, 29454–29464. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Sanefuji, M.; Morokuma, S.; Yoden, M.; Momoda, R.; Sonomoto, K.; Ogawa, M.; Kato, K.; Nakayama, J. The association between gut microbiota development and maturation of intestinal bile acid metabolism in the first 3 y of healthy Japanese infants. Gut Microbes 2020, 11, 205–216. [Google Scholar] [CrossRef]
- Sun, L.; Xie, C.; Wang, G.; Wu, Y.; Wu, Q.; Wang, X.; Liu, J.; Deng, Y.; Xia, J.; Chen, B.; et al. Gut microbiota and intestinal FXR mediate the clinical benefits of metformin. Nat. Med. 2018, 24, 1919–1929. [Google Scholar] [CrossRef]
- Zhao, L.; Chen, Y.; Xia, F.; Abudukerimu, B.; Zhang, W.; Guo, Y.; Wang, N.; Lu, Y. A glucagon-like peptide-1 receptor agonist lowers weight by modulating the structure of gut microbiota. Front. Endocrinol. 2018, 9, 233. [Google Scholar] [CrossRef] [Green Version]
- Zeng, Q.; Li, D.; He, Y.; Li, Y.; Yang, Z.; Zhao, X.; Liu, Y.; Wang, Y.; Sun, J.; Feng, X.; et al. Discrepant gut microbiota markers for the classification of obesity-related metabolic abnormalities. Sci. Rep. 2019, 9, 13424. [Google Scholar] [CrossRef] [Green Version]
- Wu, G.D.; Chen, J.; Hoffmann, C.; Bittinger, K.; Chen, Y.Y.; Keilbaugh, S.A.; Bewtra, M.; Knights, D.; Walters, W.A.; Knight, R.; et al. Linking long-term dietary patterns with gut microbial enterotypes. Science 2011, 334, 105–108. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Li, W.; Wang, C.; Wang, L.; He, T.; Hu, H.; Song, J.; Cui, C.; Qiao, J.; Qing, L.; et al. Enterotype Bacteroides is associated with a high risk in patients with diabetes: A pilot study. J Diabetes Res 2020, 2020, 6047145. [Google Scholar] [CrossRef] [Green Version]
- Durbán, A.; Abellán, J.J.; Latorre, A.; Moya, A. Effect of dietary carbohydrate restriction on an obesity-related Prevotella-dominated human fecal microbiota. Metagenomics 2013, 2, 1–4. [Google Scholar] [CrossRef]
- Chen, T.; Long, W.; Zhang, C.; Liu, S.; Zhao, L.; Hamaker, B.R. Fiber-utilizing capacity varies in Prevotella-versus Bacteroides-dominated gut microbiota. Sci. Rep. 2017, 7, 2594. [Google Scholar] [CrossRef]
- Psichas, A.; Sleeth, M.L.; Murphy, K.G.; Brooks, L.; Bewick, G.A.; Hanyaloglu, A.C.; Ghatei, M.A.; Bloom, S.R.; Frost, G. The short chain fatty acid propionate stimulates GLP-1 and PYY secretion via free fatty acid receptor 2 in rodents. Int. J. Obes. 2015, 39, 424–429. [Google Scholar] [CrossRef] [Green Version]
- De Vadder, F.; Kovatcheva-Datchary, P.; Zitoun, C.; Duchampt, A.; Bäckhed, F.; Mithieux, G. Microbiota-produced succinate improves glucose homeostasis via intestinal gluconeogenesis. Cell Metab. 2016, 24, 151–157. [Google Scholar] [CrossRef] [Green Version]
- Serena, C.; Ceperuelo-Mallafré, V.; Keiran, N.; Queipo-Ortuño, M.I.; Bernal, R.; Gomez-Huelgas, R.; Urpi-Sarda, M.; Sabater, M.; Pérez-Brocal, V.; Andrés-Lacueva, C.; et al. Elevated circulating levels of succinate in human obesity are linked to specific gut microbiota. ISME J. 2018, 12, 1642–1657. [Google Scholar] [CrossRef] [Green Version]
- Pathak, P.; Xie, C.; Nichols, R.G.; Ferrell, J.M.; Boehme, S.; Krausz, K.W.; Patterson, A.D.; Gonzalez, F.J.; Chiang, J.Y.L. Intestine farnesoid X receptor agonist and the gut microbiota activate G-protein bile acid receptor-1 signaling to improve metabolism. Hepatology 2018, 68, 1574–1588. [Google Scholar] [CrossRef]
- Wang, K.; Liao, M.; Zhou, N.; Bao, L.; Ma, K.; Zheng, Z.; Wang, Y.; Liu, C.; Wang, W.; Wang, J.; et al. Parabacteroides distasonis alleviates obesity and metabolic dysfunctions via production of succinate and secondary bile acids. Cell Rep. 2019, 26, 222–235.e5. [Google Scholar] [CrossRef] [Green Version]
- Vettorazzi, J.F.; Kurauti, M.A.; Soares, G.M.; Borck, P.C.; Ferreira, S.M.; Branco, R.C.S.; Michelone, L.D.L.; Boschero, A.C.; Junior, J.M.C.; Carneiro, E.M. Bile acid TUDCA improves insulin clearance by increasing the expression of insulin-degrading enzyme in the liver of obese mice. Sci. Rep. 2017, 7, 14876. [Google Scholar] [CrossRef] [Green Version]
- Tsuchida, T.; Shiraishi, M.; Ohta, T.; Sakai, K.; Ishii, S. Ursodeoxycholic improves insulin sensitivity and hepatic steatosis by inducing the excretion of hepatic lipids in high-fat diet-fed KK-Ay mice. Metab. Clin. Exp. 2012, 61, 944–953. [Google Scholar] [CrossRef]
- Shima, K.R.; Ota, T.; Kato, K.I.; Takeshita, Y.; Misu, H.; Kaneko, S.; Takamura, T. Ursodeoxycholic acid potentiates dipeptidyl peptidase-4 inhibitor sitagliptin by enhancing glucagon-like peptide-1 secretion in patients with type 2 diabetes and chronic liver disease: A pilot randomized controlled and add-on study. BMJ Open Diab. Res. Care 2018, 6, e000469. [Google Scholar] [CrossRef]
- Sansome, D.J.; Xie, C.; Veedfald, S.; Horowitz, M.; Rayner, C.K.; Wu, T. Mechanism of glucose-lowering by metformin in type 2 diabetes: Role of bile acids. Diabetes Obes. Metab. 2020, 22, 141–148. [Google Scholar] [CrossRef] [PubMed]
- Scarpello, J.H.B.; Hodgson, E.; Howlett, H.C.S. Effect of metformin on bile salt circulation and intestinal motility in type 2 diabetes mellitus. Diabet. Med. 1998, 15, 651–656. [Google Scholar] [CrossRef]
- Napolitano, A.; Miller, S.; Nicholls, A.W.; Baker, D.; Van Horn, S.; Thomas, E.; Rajpal, D.; Spivak, A.; Brown, J.R.; Nunez, D.J. Novel gut-based pharmacology of metformin in patients with type 2 diabetes mellitus. PLoS ONE 2014, 9, e100778. [Google Scholar] [CrossRef] [PubMed]
- Song, Z.; Cai, Y.; Lao, X.; Wang, X.; Lin, X.; Cui, Y.; Kalavagunta, P.K.; Liao, J.; Jin, L.; Shang, J.; et al. Taxonomic profiling and populational patterns of bacterial bile salt hydrolase (BSH) genes based on worldwide human gut microbiome. Microbiome 2019, 7, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Non-T2D | T2D | p Value 1 | p Value 2 | |||||
|---|---|---|---|---|---|---|---|---|
| Category: | Lean | Overweight | Obese | Lean | Overweight | Obese | ||
| No. | 25 | 7 | 18 | 11 | 11 | 3 | ||
| Gender | Male | Male | Male | Male | Male | Male | ||
| Age (years) | 47.4 ± 8.0 | 42.6 ± 2.6 | 44.2 ± 6.2 | 52.4 ± 7.7 | 52.5 ± 5.8 | 46.0 ± 7.8 | 0.0015 | 0.0824 |
| Body height (cm) | 166.2 ± 6.1 | 171.0 ± 5.4 | 167.0 ± 4.2 | 170.2 ± 5.2 | 163.1 ± 4.0 | 165.3 ± 5.5 | 0.4100 | 0.4739 |
| Body weight (kg) | 63.2 ± 7.6 | 83.7 ± 5.6 | 92.5 ± 10.9 | 63.9 ± 8.1 | 71.5 ± 3.5 | 104.0 ± 31.4 | 0.1500 | 2.31 × 10−12 |
| BMI (kg/m2) | 22.9 ± 1.7 | 28.6 ± 1.6 | 33.1 ± 2.9 | 22.0 ± 2.0 | 26.9 ± 1.5 | 37.9 ± 10.2 | 0.2700 | 1.89 × 10−14 |
| Anti-diabetic drugs (no.) | 0 | 0 | 0 | 3 | 4 | 0 | ||
| HbA1c (%) | 5.7 ± 0.3 | 5.3 ± 0.3 | 5.7 ± 0.3 | 9.6 ± 2.7 | 8.7 ± 1.3 | 7.3 ± 1.0 | 2.1 × 10−12 | 0.2961 |
| Fasting blood glucose (mg/dL) | 93.1 ± 11.9 | 89.0 ± 3.7 | 96.6 ± 10.9 | 221.5 ± 84.3 | 161.9 ± 53.8 | 138.0 ± 22.3 | 4.3 × 10−12 | 0.4084 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Therdtatha, P.; Song, Y.; Tanaka, M.; Mariyatun, M.; Almunifah, M.; Manurung, N.E.P.; Indriarsih, S.; Lu, Y.; Nagata, K.; Fukami, K.; et al. Gut Microbiome of Indonesian Adults Associated with Obesity and Type 2 Diabetes: A Cross-Sectional Study in an Asian City, Yogyakarta. Microorganisms 2021, 9, 897. https://doi.org/10.3390/microorganisms9050897
Therdtatha P, Song Y, Tanaka M, Mariyatun M, Almunifah M, Manurung NEP, Indriarsih S, Lu Y, Nagata K, Fukami K, et al. Gut Microbiome of Indonesian Adults Associated with Obesity and Type 2 Diabetes: A Cross-Sectional Study in an Asian City, Yogyakarta. Microorganisms. 2021; 9(5):897. https://doi.org/10.3390/microorganisms9050897
Chicago/Turabian StyleTherdtatha, Phatthanaphong, Yayi Song, Masaru Tanaka, Mariyatun Mariyatun, Maisaroh Almunifah, Nancy Eka Putri Manurung, Siska Indriarsih, Yi Lu, Koji Nagata, Katsuya Fukami, and et al. 2021. "Gut Microbiome of Indonesian Adults Associated with Obesity and Type 2 Diabetes: A Cross-Sectional Study in an Asian City, Yogyakarta" Microorganisms 9, no. 5: 897. https://doi.org/10.3390/microorganisms9050897