
17-AAG-Induced Activation of the Autophagic Pathway in Leishmania Is Associated with Parasite Death

 , , , and
, , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Leishmania Culturing
2.2. Generation of Parasite Mutants Expressing Fluorescent Markers
2.3. Parasite Treatment with 17-AAG or Pentamidine
2.4. Assessment of Autophagosome Formation and Autophagosome Colocalization with Glycosomes and Lysosomes by Fluorescence Microscopy
2.5. Parasite Viability
2.6. Parasite Growth Curve
2.7. Western Blot to Assess Ubiquitylated Proteins
2.8. Assessment of Protein Aggregation
2.9. Statistical Analysis
3. Results
3.1. 17-AAG Induces Autophagosome Formation in Promastigote Forms of Leishmania
3.2. 17-AAG Inhibits the Autophagosome Maturation Process
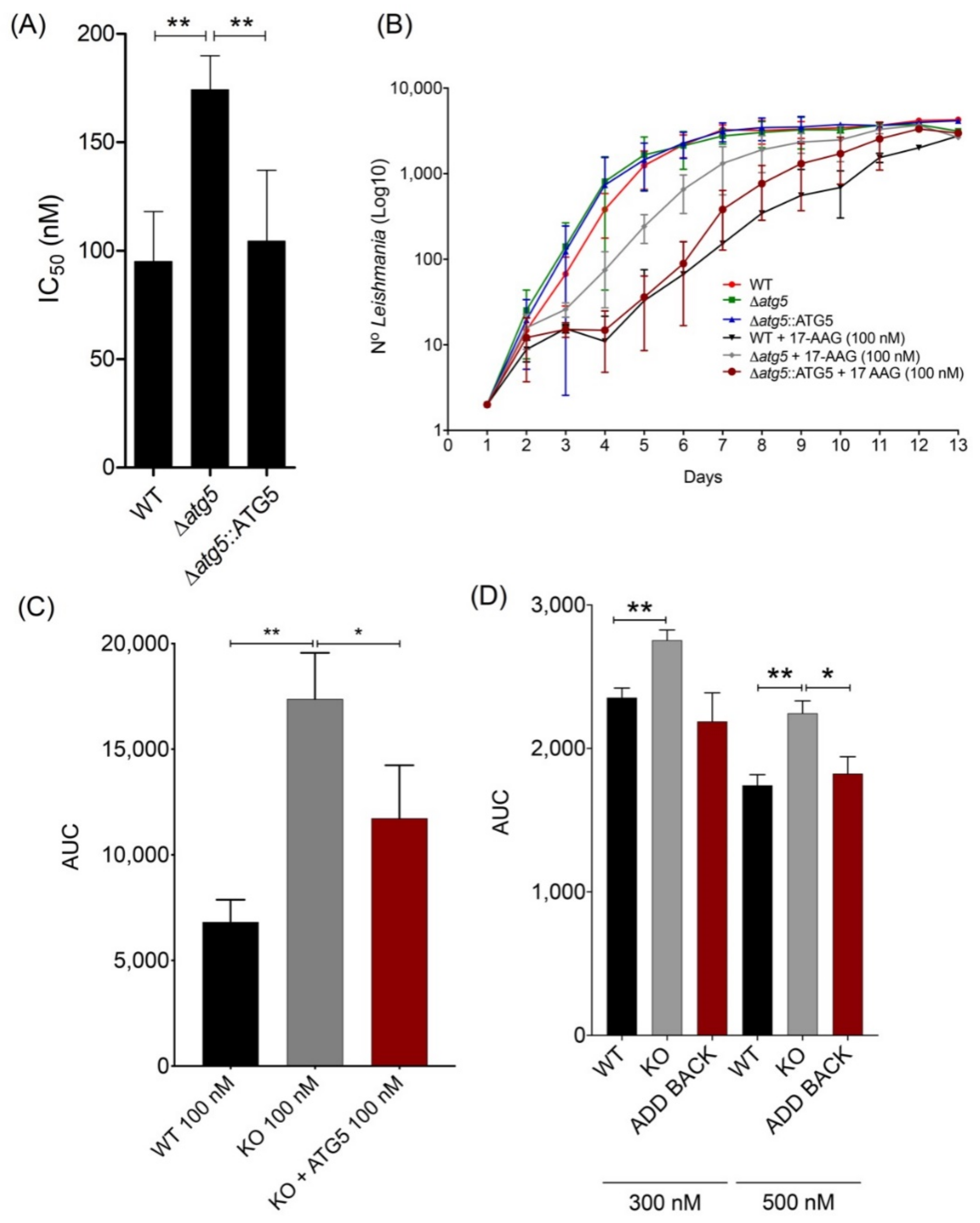
3.3. atg5-Deficient Parasites Are More Resistant to 17-AAG-Induced Cell Death Than WT Parasites
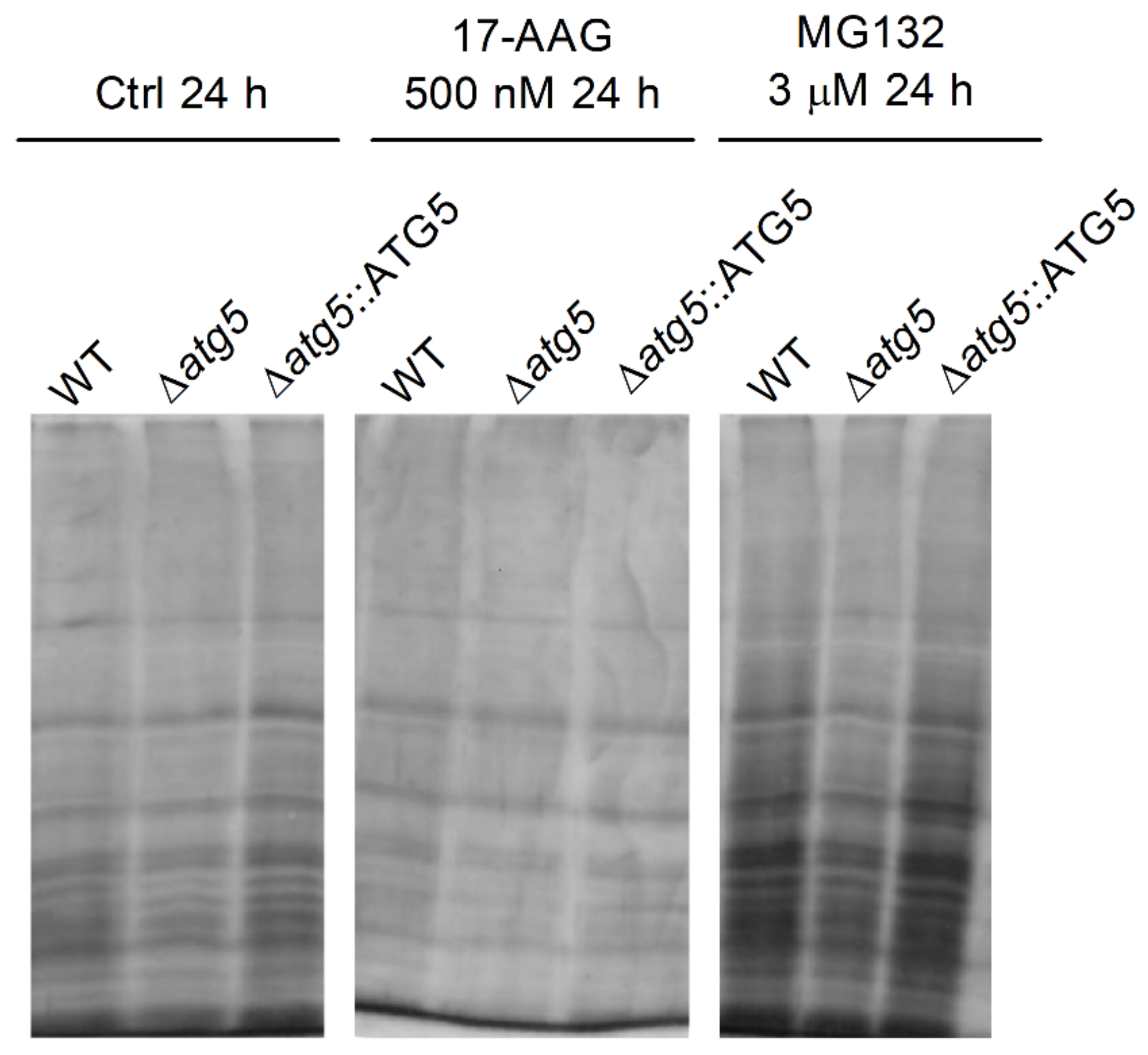
3.4. 17-AAG Treatment Results in Increased Accumulation of Ubiquitylated Proteins, But Not Protein Aggregates, in L. major Parasites
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Herwaldt, B.L. Leishmaniasis. Lancet 1999, 354, 1191–1199. [Google Scholar] [CrossRef] [Green Version]
- Banuls, A.L.; Hide, M.; Prugnolle, F. Leishmania and the leishmaniases: A parasite genetic update and advances in taxonomy, epidemiology and pathogenicity in humans. Adv. Parasitol. 2007, 64, 1–109. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Control of the Leishmaniases. World Health Organ. Tech. Rep. Ser. 2010, Xii–Xiii, 1–186. [Google Scholar]
- Oliveira, L.F.; Schubach, A.O.; Martins, M.M.; Passos, S.L.; Oliveira, R.V.; Marzochi, M.C.; Andrade, C.A. Systematic review of the adverse effects of cutaneous leishmaniasis treatment in the new world. Acta Trop. 2011, 118, 87–96. [Google Scholar] [CrossRef] [PubMed]
- Croft, S.L.; Seifert, K.; Yardley, V. Current scenario of drug development for leishmaniasis. Indian J. Med. Res. 2006, 123, 399–410. [Google Scholar] [PubMed]
- Croft, S.L.; Sundar, S.; Fairlamb, A.H. Drug resistance in leishmaniasis. Clin. Microbiol. Rev. 2006, 19, 111–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Menezes, J.P.; Guedes, C.E.; Petersen, A.L.; Fraga, D.B.; Veras, P.S. Advances in development of new treatment for leishmaniasis. BioMed Res. Int. 2015, 2015, 815023. [Google Scholar] [CrossRef]
- Angel, S.O.; Matrajt, M.; Echeverria, P.C. A review of recent patents on the protozoan parasite hsp90 as a drug target. Recent Pat. Biotechnol. 2013, 7, 2–8. [Google Scholar] [CrossRef]
- Roy, N.; Nageshan, R.K.; Ranade, S.; Tatu, U. Heat shock protein 90 from neglected protozoan parasites. Biochim. Biophys. Acta 2012, 1823, 707–711. [Google Scholar] [CrossRef] [Green Version]
- Shonhai, A.; Maier, A.G.; Przyborski, J.M.; Blatch, G.L. Intracellular protozoan parasites of humans: The role of molecular chaperones in development and pathogenesis. Protein Pept. Lett. 2011, 18, 143–157. [Google Scholar] [CrossRef] [Green Version]
- Wiesgigl, M.; Clos, J. Heat shock protein 90 homeostasis controls stage differentiation in leishmania donovani. Mol. Biol. Cell 2001, 12, 3307–3316. [Google Scholar] [CrossRef] [Green Version]
- Ahn, H.J.; Kim, S.; Nam, H.W. Molecular cloning of the 82-kda heat shock protein (hsp90) of toxoplasma gondii associated with the entry into and growth in host cells. Biochem. Biophys. Res. Commun. 2003, 311, 654–659. [Google Scholar] [CrossRef]
- Pallavi, R.; Roy, N.; Nageshan, R.K.; Talukdar, P.; Pavithra, S.R.; Reddy, R.; Venketesh, S.; Kumar, R.; Gupta, A.K.; Singh, R.K.; et al. Heat shock protein 90 as a drug target against protozoan infections: Biochemical characterization of hsp90 from plasmodium falciparum and trypanosoma evansi and evaluation of its inhibitor as a candidate drug. J. Biol. Chem. 2010, 285, 37964–37975. [Google Scholar] [CrossRef] [Green Version]
- Meyer, K.J.; Shapiro, T.A. Potent antitrypanosomal activities of heat shock protein 90 inhibitors in vitro and in vivo. J. Infect. Dis. 2013, 208, 489–499. [Google Scholar] [CrossRef] [Green Version]
- Petersen, A.L.; Guedes, C.E.; Versoza, C.L.; Lima, J.G.; de Freitas, L.A.; Borges, V.M.; Veras, P.S. 17-aag kills intracellular leishmania amazonensis while reducing inflammatory responses in infected macrophages. PLoS ONE 2012, 7, e49496. [Google Scholar] [CrossRef] [Green Version]
- Santos, D.M.; Petersen, A.L.; Celes, F.S.; Borges, V.M.; Veras, P.S.; de Oliveira, C.I. Chemotherapeutic potential of 17-aag against cutaneous leishmaniasis caused by leishmania (viannia) braziliensis. PLoS Negl. Trop. Dis. 2014, 8, e3275. [Google Scholar] [CrossRef]
- Tokui, K.; Adachi, H.; Waza, M.; Katsuno, M.; Minamiyama, M.; Doi, H.; Tanaka, K.; Hamazaki, J.; Murata, S.; Tanaka, F.; et al. 17-dmag ameliorates polyglutamine-mediated motor neuron degeneration through well-preserved proteasome function in an sbma model mouse. Hum. Mol. Genet. 2009, 18, 898–910. [Google Scholar] [CrossRef] [Green Version]
- Neckers, L.; Workman, P. Hsp90 molecular chaperone inhibitors: Are we there yet? Clin. Cancer Res. 2012, 18, 64–76. [Google Scholar] [CrossRef] [Green Version]
- Isaacs, J.S.; Xu, W.; Neckers, L. Heat shock protein 90 as a molecular target for cancer therapeutics. Cancer Cell 2003, 3, 213–217. [Google Scholar] [CrossRef] [Green Version]
- Zuehlke, A.; Johnson, J.L. Hsp90 and co-chaperones twist the functions of diverse client proteins. Biopolymers 2010, 93, 211–217. [Google Scholar] [CrossRef]
- Driscoll, J.J.; Chowdhury, R.D. Molecular crosstalk between the proteasome, aggresomes and autophagy: Translational potential and clinical implications. Cancer Lett. 2012, 325, 147–154. [Google Scholar] [CrossRef]
- Wolff, S.; Weissman, J.S.; Dillin, A. Differential scales of protein quality control. Cell 2014, 157, 52–64. [Google Scholar] [CrossRef] [Green Version]
- Tyedmers, J.; Mogk, A.; Bukau, B. Cellular strategies for controlling protein aggregation. Nat. Rev. Mol. Cell Biol. 2010, 11, 777–788. [Google Scholar] [CrossRef]
- Sittler, A.; Lurz, R.; Lueder, G.; Priller, J.; Lehrach, H.; Hayer-Hartl, M.K.; Hartl, F.U.; Wanker, E.E. Geldanamycin activates a heat shock response and inhibits huntingtin aggregation in a cell culture model of huntington’s disease. Hum. Mol. Genet. 2001, 10, 1307–1315. [Google Scholar] [CrossRef] [Green Version]
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of cells and tissues. Cell 2011, 147, 728–741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, Y.; Ren, X.; Hait, W.N.; Yang, J.M. Therapeutic targeting of autophagy in disease: Biology and pharmacology. Pharmacol. Rev. 2013, 65, 1162–1197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klionsky, D.J.; Emr, S.D. Autophagy as a regulated pathway of cellular degradation. Science 2000, 290, 1717–1721. [Google Scholar] [CrossRef] [PubMed]
- Yorimitsu, T.; Klionsky, D.J. Eating the endoplasmic reticulum: Quality control by autophagy. Trends Cell Biol. 2007, 17, 279–285. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Klionsky, D.J. Protein turnover via autophagy: Implications for metabolism. Annu. Rev. Nutr. 2007, 27, 19–40. [Google Scholar] [CrossRef] [Green Version]
- Klionsky, D.J.; Abdalla, F.C.; Abeliovich, H.; Abraham, R.T.; Acevedo-Arozena, A.; Adeli, K.; Agholme, L.; Agnello, M.; Agostinis, P.; Aguirre-Ghiso, J.A.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 2012, 8, 445–544. [Google Scholar] [CrossRef]
- Duszenko, M.; Ginger, M.L.; Brennand, A.; Gualdron-Lopez, M.; Colombo, M.I.; Coombs, G.H.; Coppens, I.; Jayabalasingham, B.; Langsley, G.; de Castro, S.L.; et al. Autophagy in protists. Autophagy 2011, 7, 127–158. [Google Scholar] [CrossRef] [Green Version]
- Meijer, W.H.; van der Klei, I.J.; Veenhuis, M.; Kiel, J.A. Atg genes involved in non-selective autophagy are conserved from yeast to man, but the selective cvt and pexophagy pathways also require organism-specific genes. Autophagy 2007, 3, 106–116. [Google Scholar] [CrossRef] [Green Version]
- Besteiro, S.; Williams, R.A.; Morrison, L.S.; Coombs, G.H.; Mottram, J.C. Endosome sorting and autophagy are essential for differentiation and virulence of leishmania major. J. Biol. Chem. 2006, 281, 11384–11396. [Google Scholar] [CrossRef] [Green Version]
- Besteiro, S.; Williams, R.A.; Coombs, G.H.; Mottram, J.C. Protein turnover and differentiation in leishmania. Int. J. Parasitol. 2007, 37, 1063–1075. [Google Scholar] [CrossRef] [Green Version]
- Williams, R.A.; Smith, T.K.; Cull, B.; Mottram, J.C.; Coombs, G.H. Atg5 is essential for atg8-dependent autophagy and mitochondrial homeostasis in leishmania major. PLoS Pathog 2012, 8, e1002695. [Google Scholar] [CrossRef] [Green Version]
- Meijer, A.J.; Codogno, P. Regulation and role of autophagy in mammalian cells. Int. J. Biochem. Cell Biol. 2004, 36, 2445–2462. [Google Scholar] [CrossRef]
- Levine, B.; Klionsky, D.J. Development by self-digestion: Molecular mechanisms and biological functions of autophagy. Dev. Cell 2004, 6, 463–477. [Google Scholar] [CrossRef]
- Levine, B.; Yuan, J. Autophagy in cell death: An innocent convict? J. Clin. Investig. 2005, 115, 2679–2688. [Google Scholar] [CrossRef]
- Sengupta, S.; Chowdhury, S.; Bosedasgupta, S.; Wright, C.W.; Majumder, H.K. Cryptolepine-induced cell death of leishmania donovani promastigotes is augmented by inhibition of autophagy. Mol. Biol. Int. 2011, 2011, 187850. [Google Scholar] [CrossRef] [Green Version]
- Cull, B.; Prado Godinho, J.L.; Fernandes Rodrigues, J.C.; Frank, B.; Schurigt, U.; Williams, R.A.; Coombs, G.H.; Mottram, J.C. Glycosome turnover in leishmania major is mediated by autophagy. Autophagy 2014, 10, 2143–2157. [Google Scholar] [CrossRef] [Green Version]
- Huete-Perez, J.A.; Engel, J.C.; Brinen, L.S.; Mottram, J.C.; McKerrow, J.H. Protease trafficking in two primitive eukaryotes is mediated by a prodomain protein motif. J. Biol. Chem. 1999, 274, 16249–16256. [Google Scholar] [CrossRef] [Green Version]
- Williams, R.A.; Woods, K.L.; Juliano, L.; Mottram, J.C.; Coombs, G.H. Characterization of unusual families of atg8-like proteins and atg12 in the protozoan parasite leishmania major. Autophagy 2009, 5, 159–172. [Google Scholar] [CrossRef] [Green Version]
- Bolte, S.; Cordelieres, F.P. A guided tour into subcellular colocalization analysis in light microscopy. J. Microsc. 2006, 224, 213–232. [Google Scholar] [CrossRef]
- Jang, H.H.; Lee, K.O.; Chi, Y.H.; Jung, B.G.; Park, S.K.; Park, J.H.; Lee, J.R.; Lee, S.S.; Moon, J.C.; Yun, J.W.; et al. Two enzymes in one; two yeast peroxiredoxins display oxidative stress-dependent switching from a peroxidase to a molecular chaperone function. Cell 2004, 117, 625–635. [Google Scholar] [CrossRef]
- Amaravadi, R.K.; Thompson, C.B. The roles of therapy-induced autophagy and necrosis in cancer treatment. Clin. Cancer Res. 2007, 13, 7271–7279. [Google Scholar] [CrossRef] [Green Version]
- Cervantes, S.; Bunnik, E.M.; Saraf, A.; Conner, C.M.; Escalante, A.; Sardiu, M.E.; Ponts, N.; Prudhomme, J.; Florens, L.; Le Roch, K.G. The multifunctional autophagy pathway in the human malaria parasite, plasmodium falciparum. Autophagy 2014, 10, 80–92. [Google Scholar] [CrossRef] [Green Version]
- Hoppe, H.C.; van Schalkwyk, D.A.; Wiehart, U.I.; Meredith, S.A.; Egan, J.; Weber, B.W. Antimalarial quinolines and artemisinin inhibit endocytosis in plasmodium falciparum. Antimicrob. Agents Chemother. 2004, 48, 2370–2378. [Google Scholar] [CrossRef] [Green Version]
- Akerfelt, M.; Morimoto, R.I.; Sistonen, L. Heat shock factors: Integrators of cell stress, development and lifespan. Nat. Rev. Mol. Cell Biol. 2010, 11, 545–555. [Google Scholar] [CrossRef]
- Hartl, F.U. Molecular chaperones in cellular protein folding. Nature 1996, 381, 571–579. [Google Scholar] [CrossRef]
- Hartl, F.U.; Bracher, A.; Hayer-Hartl, M. Molecular chaperones in protein folding and proteostasis. Nature 2011, 475, 324–332. [Google Scholar] [CrossRef]
- Chen, C.Y.; Balch, W.E. The hsp90 chaperone complex regulates gdi-dependent rab recycling. Mol. Biol. Cell 2006, 17, 3494–3507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lotz, G.P.; Brychzy, A.; Heinz, S.; Obermann, W.M. A novel hsp90 chaperone complex regulates intracellular vesicle transport. J. Cell Sci. 2008, 121, 717–723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Cristina, M.; Dou, Z.; Lunghi, M.; Kannan, G.; Huynh, M.H.; McGovern, O.L.; Schultz, T.L.; Schultz, A.J.; Miller, A.J.; Hayes, B.M.; et al. Toxoplasma depends on lysosomal consumption of autophagosomes for persistent infection. Nat. Microbiol. 2017, 2, 17096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, A.M.; Ryter, S.W.; Levine, B. Autophagy in human health and disease. N. Engl. J. Med. 2013, 368, 651–662. [Google Scholar] [CrossRef]
- Degenhardt, K.; Mathew, R.; Beaudoin, B.; Bray, K.; Anderson, D.; Chen, G.; Mukherjee, C.; Shi, Y.; Gelinas, C.; Fan, Y.; et al. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell 2006, 10, 51–64. [Google Scholar] [CrossRef] [Green Version]
- Lum, J.J.; Bauer, D.E.; Kong, M.; Harris, M.H.; Li, C.; Lindsten, T.; Thompson, C.B. Growth factor regulation of autophagy and cell survival in the absence of apoptosis. Cell 2005, 120, 237–248. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, H.M.; Berry, L.; Sullivan, W.J., Jr.; Besteiro, S. Autophagy participates in the unfolded protein response in toxoplasma gondii. FEMS Microbiol. Lett. 2017, 364. [Google Scholar] [CrossRef]
- Mizushima, N.; Levine, B.; Cuervo, A.M.; Klionsky, D.J. Autophagy fights disease through cellular self-digestion. Nature 2008, 451, 1069–1075. [Google Scholar] [CrossRef] [Green Version]
- Chung, Y.; Lee, J.; Jung, S.; Lee, Y.; Cho, J.W.; Oh, Y.J. Dysregulated autophagy contributes to caspase-dependent neuronal apoptosis. Cell Death Dis. 2018, 9, 1189. [Google Scholar] [CrossRef] [Green Version]
- Williams, R.A.; Tetley, L.; Mottram, J.C.; Coombs, G.H. Cysteine peptidases cpa and cpb are vital for autophagy and differentiation in leishmania mexicana. Mol. Microbiol. 2006, 61, 655–674. [Google Scholar] [CrossRef]
- Theodoraki, M.A.; Caplan, A.J. Quality control and fate determination of hsp90 client proteins. Biochim. Biophys. Acta 2012, 1823, 683–688. [Google Scholar] [CrossRef] [Green Version]
- Janen, S.B.; Chaachouay, H.; Richter-Landsberg, C. Autophagy is activated by proteasomal inhibition and involved in aggresome clearance in cultured astrocytes. Glia 2010, 58, 1766–1774. [Google Scholar] [CrossRef]
- Grumati, P.; Dikic, I. Ubiquitin signaling and autophagy. J. Biol. Chem. 2018, 293, 5404–5413. [Google Scholar] [CrossRef] [Green Version]
- Grumati, P.; Morozzi, G.; Holper, S.; Mari, M.; Harwardt, M.I.; Yan, R.; Muller, S.; Reggiori, F.; Heilemann, M.; Dikic, I. Full length rtn3 regulates turnover of tubular endoplasmic reticulum via selective autophagy. Elife 2017, 6. [Google Scholar] [CrossRef]
- Silva-Fernandes, A.; Duarte-Silva, S.; Neves-Carvalho, A.; Amorim, M.; Soares-Cunha, C.; Oliveira, P.; Thirstrup, K.; Teixeira-Castro, A.; Maciel, P. Chronic treatment with 17-dmag improves balance and coordination in a new mouse model of machado-joseph disease. Neurotherapeutics 2014, 11, 433–449. [Google Scholar] [CrossRef] [Green Version]
- Bose, S.; Cho, J. Targeting chaperones, heat shock factor-1, and unfolded protein response: Promising therapeutic approaches for neurodegenerative disorders. Ageing Res. Rev. 2017, 35, 155–175. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Petersen, A.L.d.O.A.; Cull, B.; Dias, B.R.S.; Palma, L.C.; Luz, Y.d.S.; de Menezes, J.P.B.; Mottram, J.C.; Veras, P.S.T. 17-AAG-Induced Activation of the Autophagic Pathway in Leishmania Is Associated with Parasite Death. Microorganisms 2021, 9, 1089. https://doi.org/10.3390/microorganisms9051089
Petersen ALdOA, Cull B, Dias BRS, Palma LC, Luz YdS, de Menezes JPB, Mottram JC, Veras PST. 17-AAG-Induced Activation of the Autophagic Pathway in Leishmania Is Associated with Parasite Death. Microorganisms. 2021; 9(5):1089. https://doi.org/10.3390/microorganisms9051089
Chicago/Turabian StylePetersen, Antonio Luis de O. A., Benjamin Cull, Beatriz R. S. Dias, Luana C. Palma, Yasmin da S. Luz, Juliana P. B. de Menezes, Jeremy C. Mottram, and Patrícia S. T. Veras. 2021. "17-AAG-Induced Activation of the Autophagic Pathway in Leishmania Is Associated with Parasite Death" Microorganisms 9, no. 5: 1089. https://doi.org/10.3390/microorganisms9051089