
Physiological and Genomic Analysis of Bacillus pumilus UAMX Isolated from the Gastrointestinal Tract of Overweight Individuals

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Strain Propagation
2.2. GenBank Accession Number for the Studied Strain
2.3. Resistance Assessment to Simulated GIT Physiological Conditions
2.4. Growth Ability in Different Carbon Sources
2.5. Electrophoretic Analysis of BP-UAMX Cultured with Different Carbon Sources
2.6. Proteomic Profiling Analysis for BP-UAMX
2.7. Sequencing and Total Genome Assembly
2.8. Gene Grouping by Function and Metabolic Pathway Prediction
2.9. Pan-Genome Analysis
2.10. Statistical Analysis
3. Results and Discussion
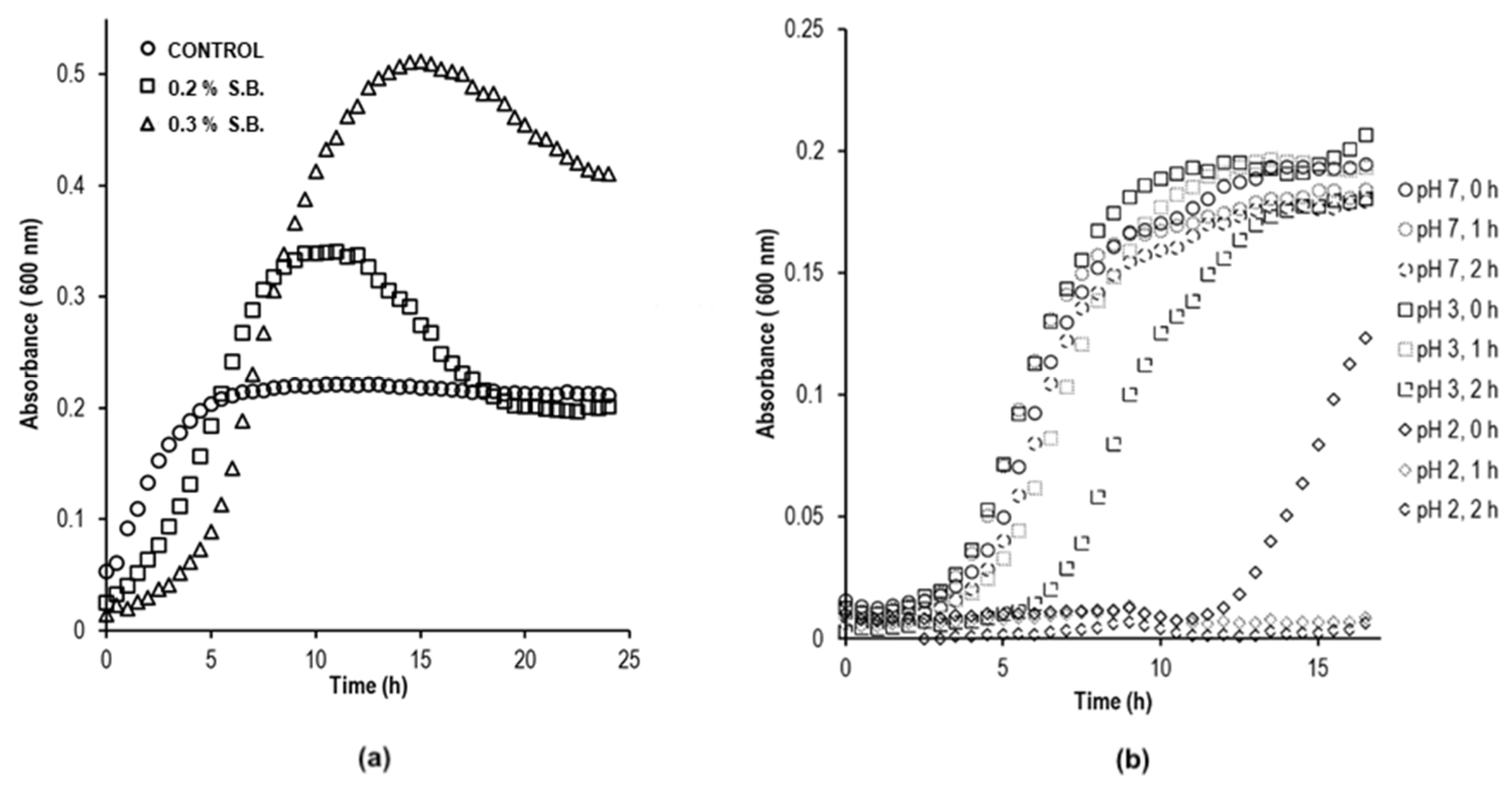
3.1. Resistance to Gastrointestinal Conditions
3.2. Analysis of Growth Kinetics Using Different Carbon Sources
3.3. Electrophoretic Analysis of BP-UAMX Cultured with Different Carbon Sources
3.4. Genome Sequencing and Assembly
3.5. Pan-Genome Analysis
3.6. Gene Grouping by Function and Metabolic Pathway Prediction
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fakhry, S.; Sorrentini, I.; Ricca, E.; De Felice, M.; Baccigalupi, L. Characterization of spore forming Bacilli isolated from the human gastrointestinal tract. J. Appl. Microbiol. 2008, 105, 2178–2186. [Google Scholar] [CrossRef]
- Hong, H.A.; To, E.; Fakhry, S.; Baccigalupi, L.; Ricca, E.; Cutting, S.M. Defining the natural habitat of Bacillus spore-formers. Res. Microbiol. 2009, 160, 375–379. [Google Scholar] [CrossRef]
- Rajilić-Stojanović, M.; De Vos, W.M. The first 1000 cultured species of the human gastrointestinal microbiota. FEMS Microbiol. Rev. 2014, 38, 996–1047. [Google Scholar] [CrossRef]
- Reyes, L.M.; Vázquez, R.G.; Arroyo, S.M.C.; Avalos, A.M.; Castillo, P.A.R.; Pérez, D.A.C.; Terrones, I.R.; Ibáñez, N.R.; Magallanes, M.M.R.; Langella, P.; et al. Correlation between diet and gut bacteria in a population of young adults. Int. J. Food Sci. Nutr. 2016, 67, 470–478. [Google Scholar] [CrossRef]
- Alou, M.T.; Fournier, P.-E.; Raoult, D. “Bacillus mediterraneensis”, a new bacterial species isolated from human gut microbiota. New Microbes New Infect. 2016, 12, 86–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopetuso, L.R.; Scaldaferri, F.; Franceschi, F.; Gasbarrini, A. Bacillus clausii and gut homeostasis: State of the art and future perspectives. Expert Rev. Gastroenterol. Hepatol. 2016, 10, 943–948. [Google Scholar] [CrossRef]
- Allen, A. Structure and function of gastrointestinal mucus. In Physiology of the Gastrointestinal Tract; Johnson, L.R., Ed.; Raven Press: New York, NY, USA, 1981; Chapter 22. [Google Scholar] [CrossRef]
- Charteris, W.P.; Kelly, P.M.; Morelli, L.; Collins, J.K. Development and application of an In Vitro methodology to determine the transit tolerance of potentially probiotic Lactobacillus and Bifidobacterium species in the upper human gastrointestinal tract. J. Appl. Microbiol. 1998, 84, 759–768. [Google Scholar] [CrossRef]
- Tam, N.K.M.; Uyen, N.Q.; Hong, H.A.; Duc, L.H.; Hoa, T.T.; Serra, C.R.; Henriques, A.O.; Cutting, S.M. The Intestinal Life Cycle of Bacillus subtilis and Close Relatives. J. Bacteriol. 2006, 188, 2692–2700. [Google Scholar] [CrossRef] [Green Version]
- Ghelardi, E.; Celandroni, F.; Salvetti, S.; Gueye, S.; Lupetti, A.; Senesi, S. Survival and persistence of Bacillus clausii in the human gastrointestinal tract following oral administration as spore-based probiotic formulation. J. Appl. Microbiol. 2015, 119, 552–559. [Google Scholar] [CrossRef]
- Mingmongkolchai, S.; Panbangred, W. Bacillusprobiotics: An alternative to antibiotics for livestock production. J. Appl. Microbiol. 2018, 124, 1334–1346. [Google Scholar] [CrossRef]
- Marzorati, M.; Abbeele, P.V.D.; Bubeck, S.S.; Bayne, T.; Krishnan, K.; Young, A.; Mehta, D.; DeSouza, A. Bacillus subtilis HU58 and Bacillus coagulans SC208 Probiotics Reduced the Effects of Antibiotic-Induced Gut Microbiome Dysbiosis in An M-SHIME® Model. Microorganisms 2020, 8, 1028. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.-S.; Lee, Y.-J.; Tsai, K.-N.; Ho, Y.-H.; Fang, S.-B. Probiotic Cocktail Identified by Microbial Network Analysis Inhibits Growth, Virulence Gene Expression, and Host Cell Colonization of Vancomycin-Resistant Enterococci. Microorganisms 2020, 8, 816. [Google Scholar] [CrossRef]
- Lee, N.-K.; Kim, W.-S.; Paik, H.-D. Bacillus strains as human probiotics: Characterization, safety, microbiome, and probiotic carrier. Food Sci. Biotechnol. 2019, 28, 1297–1305. [Google Scholar] [CrossRef]
- Elshaghabee, F.M.F.; Rokana, N.; Gulhane, R.D.; Sharma, C.; Panwar, H. Bacillus As Potential Probiotics: Status, Concerns, and Future Perspectives. Front. Microbiol. 2017, 8, 1490. [Google Scholar] [CrossRef] [Green Version]
- Vo, T.T.-T.; Park, J.-H. Characteristics of Potential Gamma-Aminobutyric Acid-Producing Bacteria Isolated from Korean and Vietnamese Fermented Fish Products. J. Microbiol. Biotechnol. 2019, 29, 209–221. [Google Scholar] [CrossRef] [Green Version]
- Lee, N.-K.; Son, S.-H.; Jeon, E.B.; Jung, G.H.; Lee, J.-Y.; Paik, H.-D. The prophylactic effect of probiotic Bacillus polyfermenticus KU3 against cancer cells. J. Funct. Foods 2015, 14, 513–518. [Google Scholar] [CrossRef]
- Nannan, C.; Vu, H.Q.; Gillis, A.; Caulier, S.; Nguyen, T.T.T.; Mahillon, J. Bacilysin within the Bacillus subtilis group: Gene prevalence versus antagonistic activity against Gram-negative foodborne pathogens. J. Biotechnol. 2021, 327, 28–35. [Google Scholar] [CrossRef] [PubMed]
- De Angelis, M.; Siragusa, S.; Vacca, M.; Di Cagno, R.; Cristofori, F.; Schwarm, M.; Pelzer, S.; Flügel, M.; Speckmann, B.; Francavilla, R.; et al. Selection of Gut-Resistant Bacteria and Construction of Microbial Consortia for Improving Gluten Digestion under Simulated Gastrointestinal Conditions. Nutrients 2021, 13, 992. [Google Scholar] [CrossRef]
- Jang, C.; Oh, J.; Lim, J.; Kim, H.; Kim, J.-S. Fermented Soy Products: Beneficial Potential in Neurodegenerative Diseases. Foods 2021, 10, 636. [Google Scholar] [CrossRef]
- Bai, L.; Gao, M.; Cheng, X.; Kang, G.; Cao, X.; Huang, H. Engineered butyrate-producing bacteria prevents high fat diet-induced obesity in mice. Microb. Cell Fact. 2020, 19, 94. [Google Scholar] [CrossRef]
- Turnbaugh, P.J.; Ley, R.E.; Mahowald, M.A.; Magrini, V.; Mardis, E.R.; Gordon, J.I. An obesity-associated gut microbiome with increased capacity for energy harvest. Nat. Cell Biol. 2006, 444, 1027–1031. [Google Scholar] [CrossRef]
- Blaut, M. Gut microbiota and energy balance: Role in obesity. Proc. Nutr. Soc. 2015, 74, 227–234. [Google Scholar] [CrossRef] [Green Version]
- Leser, T.; Knarreborg, A.; Worm, J. Germination and outgrowth of Bacillus subtilis and Bacillus licheniformis spores in the gastrointestinal tract of pigs. J. Appl. Microbiol. 2008, 104, 1025–1033. [Google Scholar] [CrossRef] [PubMed]
- Pop, M. Genome assembly reborn: Recent computational challenges. Brief. Bioinform. 2009, 10, 354–366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marchesi, J.R. Human distal gut microbiome. Environ. Microbiol. 2011, 13, 3088–3102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narzisi, G.; Mishra, B. Comparing De Novo Genome Assembly: The Long and Short of It. PLoS ONE 2011, 6, e19175. [Google Scholar] [CrossRef]
- Rodríguez-Santiago, B.; Armengol, L. Tecnologías de secuenciación de nueva generación en diagnóstico genético pre-y postnatal. Diagn. Prenat. 2012, 23, 56–66. [Google Scholar] [CrossRef]
- Aguilar-Bultet, L.; Falquet, L. Secuenciación y ensamblaje de novo de genomas bacterianos: Una alternativa para el estudio de nuevos patógenos. Rev. Salud. Anim. 2015, 37, 125–132. [Google Scholar]
- Rhoads, A.; Au, K.F. PacBio Sequencing and Its Applications. Genom. Proteom. Bioinform. 2015, 13, 278–289. [Google Scholar] [CrossRef] [Green Version]
- Tatusov, R.L.; Natale, D.A.; Garkavtsev, I.V.; Tatusova, T.A.; Shankavaram, U.T.; Rao, B.S.; Kiryutin, B.; Galperin, M.Y.; Fedorova, N.D.; Koonin, E.V. The COG database: New developments in phylogenetic classification of proteins from complete genomes. Nucleic Acids Res. 2001, 29, 22–28. [Google Scholar] [CrossRef]
- Li, L.; Stoeckert, C.J.; Roos, D.S. OrthoMCL: Identification of Ortholog Groups for Eukaryotic Genomes. Genome Res. 2003, 13, 2178–2189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- KAAS-KEGG Automatic Annotation Server. Available online: https://www.genome.jp/kegg/kaas/ (accessed on 6 May 2021).
- Moriya, Y.; Itoh, M.; Okuda, S.; Yoshizawa, A.C.; Kanehisa, M. KAAS: An automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 2007, 35, W182–W185. [Google Scholar] [CrossRef] [Green Version]
- Tokimatsu, T.; Kotera, M.; Goto, S.; Kanehisa, M. KEGG and GenomeNet Resources for Predicting Protein Function from Omics Data Including KEGG PLANT Resource. In Protein Function Prediction for Omics Era; Springer: Dordrecht, The Netherlands, 2011; pp. 271–288. [Google Scholar] [CrossRef]
- Medini, D.; Donati, C.; Tettelin, H.; Masignani, V.; Rappuoli, R. The microbial pan-genome. Curr. Opin. Genet. Dev. 2005, 15, 589–594. [Google Scholar] [CrossRef]
- Page, A.J.; Cummins, C.A.; Hunt, M.; Wong, V.K.; Reuter, S.; Holden, M.T.G.; Fookes, M.; Falush, D.; Keane, J.A.; Parkhill, J. Roary: Rapid large-scale prokaryote pan genome analysis. Bioinformatics 2015, 31, 3691–3693. [Google Scholar] [CrossRef] [PubMed]
- Vernikos, G.; Medini, D.; Riley, D.R.; Tettelin, H. Ten years of pan-genome analyses. Curr. Opin. Microbiol. 2015, 23, 148–154. [Google Scholar] [CrossRef]
- Hernández-Alcántara, A.M.; Wacher, C.; Llamas, M.G.; López, P.; Pérez-Chabela, M.L. Probiotic properties and stress response of thermotolerant lactic acid bacteria isolated from cooked meat products. Int. Food Res. J. 2018, 91, 249–257. [Google Scholar] [CrossRef]
- Gatto, M.; Muratori, S.; Rinaldi, S. A functional interpretation of the logistic equation. Ecol. Model. 1988, 42, 155–159. [Google Scholar] [CrossRef]
- Hintze, J.L. User’s Guide V. Available online: https://www.ncss.com/wp-content/uploads/2012/09/NCSSUG5.pdf (accessed on 12 April 2021).
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-Dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Laemmli, U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970, 227, 680–685. [Google Scholar] [CrossRef]
- Pérez-Acosta, J.A.; Martínez-Porchas, M.; Elizalde-Contreras, J.M.; Leyva, J.M.; Ruiz-May, E.; Gollas-Galván, T.; Martínez-Córdova, L.R.; Huerta-Ocampo, J.Á. Proteomic profiling of integral membrane proteins. Microbiol. Immunol. 2018, 62, 14–23. [Google Scholar] [CrossRef] [Green Version]
- Tatusov, R.L.; Galperin, M.Y.; Natale, D.A.; Koonin, E.V. The COG database: A tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res. 2000, 28, 33–36. [Google Scholar] [CrossRef] [Green Version]
- Aoki-Kinoshita, K.F. Overview of KEGG applications to omics-related research. J. Pestic. Sci. 2006, 31, 296–299. [Google Scholar] [CrossRef] [Green Version]
- Koren, S.; Walenz, B.P.; Berlin, K.; Miller, J.R.; Bergman, N.H.; Phillippy, A.M. Canu: Scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 2017, 27, 722–736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wick, R.R.; Judd, L.M.; Gorrie, C.L.; Holt, K.E. Unicycler: Resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput. Biol. 2017, 13, e1005595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality assessment tool for genome assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef] [PubMed]
- Seemann, T. Prokka: Rapid Prokaryotic Genome Annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef]
- Contreras-Moreira, B.; Vinuesa, P. GET_HOMOLOGUES, a Versatile Software Package for Scalable and Robust Microbial Pangenome Analysis. Appl. Environ. Microbiol. 2013, 79, 7696–7701. [Google Scholar] [CrossRef] [Green Version]
- Vinuesa, P.; Ochoa-Sánchez, L.E.; Contreras-Moreira, B. GET_PHYLOMARKERS, a Software Package to Select Optimal Orthologous Clusters for Phylogenomics and Inferring Pan-Genome Phylogenies, Used for a Critical Geno-Taxonomic Revision of the Genus Stenotrophomonas. Front. Microbiol. 2018, 9, 771. [Google Scholar] [CrossRef] [Green Version]
- NCSS 2007 Update (Version 1). Available online: https://www.ncss.com/download/ncss/updates/ncss-2007-v1/ (accessed on 17 January 2021).
- Villarreal, M.L.M.; Padilha, M.; Vieira, A.D.S.; Franco, B.D.G.D.M.; Martinez, R.C.R.; Saad, S.M.I. Advantageous Direct Quantification of Viable Closely Related Probiotics in Petit-Suisse Cheeses under In Vitro Gastrointestinal Conditions by Propidium Monoazide-qPCR. PLoS ONE 2013, 8, e82102. [Google Scholar] [CrossRef]
- Li, C.; Yu, W.; Wu, P.; Chen, X.D. Current in vitro digestion systems for understanding food digestion in human upper gastrointestinal tract. Trends Food Sci. Technol. 2020, 96, 114–126. [Google Scholar] [CrossRef]
- Stasiak-Różańska, L.; Berthold-Pluta, A.; Pluta, A.; Dasiewicz, K.; Garbowska, M. Effect of Simulated Gastrointestinal Tract Conditions on Survivability of Probiotic Bacteria Present in Commercial Preparations. Int. J. Environ. Res. Public Health 2021, 18, 1108. [Google Scholar] [CrossRef]
- Veisseire, P.; Bonnet, M.; Saraoui, T.; Poupet, C.; Camarès, O.; Gachinat, M.; Callon, C.; Febvre, G.; Chassard, C.; Bornes, S. Investigation into In Vitro and In Vivo Caenorhabditis elegans Models to Select Cheese Yeasts as Probiotic Candidates for their Preventive Effects against Salmonella Typhimurium. Microorganisms 2020, 8, 922. [Google Scholar] [CrossRef]
- Kristoffersen, S.M.; Ravnum, S.; Tourasse, N.J.; Økstad, O.A.; Kolstø, A.B.; Davies, W. Low concentrations of bile salts induce stress responses and reduce motility in Bacillus cereus ATCC 14570. J. Bacteriol. 2007, 189, 5302–5313. [Google Scholar] [CrossRef] [Green Version]
- Dressman, J.B.; Berardi, R.R.; Dermentzoglou, L.C.; Russell, T.L.; Schmaltz, S.P.; Barnett, J.L.; Jarvenpaa, K.M. Upper Gastrointestinal (GI) pH in Young, Healthy Men and Women. Pharm. Res. 1990, 7, 756–761. [Google Scholar] [CrossRef]
- Berthold-Pluta, A.; Pluta, A.; Garbowska, M. The effect of selected factors on the survival of Bacillus cereus in the human gastrointestinal tract. Microb. Pathog. 2015, 82, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Soares, M.B.; Martinez, R.C.; Pereira, E.P.; Balthazar, C.F.; Cruz, A.G.; Ranadheera, C.S.; Sant’Ana, A.S. The resistance of Bacillus, Bifidobacterium, and Lactobacillus strains with claimed probiotic properties in different food matrices exposed to simulated gastrointestinal tract conditions. Food Res. Int. 2019, 125, 108542. [Google Scholar] [CrossRef] [PubMed]
- Dartois, V.; Coppée, J.Y.; Colson, C.; Baulard, A. Genetic analysis and overexpression of lipolytic activity in Bacillus subtilis. Appl. Environ. Microbiol. 1994, 60, 1670–1673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eggert, T.; Van Pouderoyen, G.; Dijkstra, B.W.; Jaeger, K.-E. Lipolytic enzymes LipA and LipB from Bacillus subtilis differ in regulation of gene expression, biochemical properties, and three-dimensional structure. FEBS Lett. 2001, 502, 89–92. [Google Scholar] [CrossRef] [Green Version]
- Ertuğrul, S.; Dönmez, G.; Takaç, S. Isolation of lipase producing Bacillus sp. from olive mill wastewater and improving its enzyme activity. J. Hazard. Mater. 2007, 149, 720–724. [Google Scholar] [CrossRef]
- Mallozzi, M.; Viswanathan, V.; Vedantam, G. Spore-forming Bacilli and Clostridia in human disease. Future Microbiol. 2010, 5, 1109–1123. [Google Scholar] [CrossRef]
- Handtke, S.; Albrecht, D.; Otto, A.; Becher, D.; Hecker, M.; Voigt, B. The Proteomic Response of Bacillus pumilus Cells to Glucose Starvation. Proteomics 2018, 18, 1700109. [Google Scholar] [CrossRef]
- Lima-Pérez, J.; López-Pérez, M.; Viniegra-González, G.; Loera, O. Solid-state fermentation of Bacillus thuringiensis var kurstaki HD-73 maintains higher biomass and spore yields as compared to submerged fermentation using the same media. Bioprocess Biosyst. Eng. 2019, 42, 1527–1535. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhya, I.; Moraïs, S.; Laverde-Gomez, J.; Sheridan, P.O.; Walker, A.W.; Kelly, W.; Klieve, A.V.; Ouwerkerk, D.; Duncan, S.H.; Louis, P.; et al. Sporulation capability and amylosome conservation among diverse human colonic and rumen isolates of the keystone starch-degrader Ruminococcus bromii. Environ. Microbiol. 2018, 20, 324–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrer, M.; Ruiz, A.; Lanza, F.; Haange, S.-B.; Oberbach, A.; Till, H.; Bargiela, R.; Campoy, C.; Segura, M.T.; Richter, M.; et al. Microbiota from the distal guts of lean and obese adolescents exhibit partial functional redundancy besides clear differences in community structure. Environ. Microbiol. 2013, 15, 211–226. [Google Scholar] [CrossRef]
- Koburger, T.; Weibezahn, J.; Bernhardt, J.; Homuth, G.; Hecker, M. Genome-wide mRNA profiling in glucose starved Bacillus subtilis cells. Mol. Genet. Genom. 2005, 274, 1–12. [Google Scholar] [CrossRef]
- Voigt, B.; Hoi, L.T.; Jürgen, B.; Albrecht, D.; Ehrenreich, A.; Veith, B.; Evers, S.; Maurer, K.-H.; Hecker, M.; Schweder, T. The glucose and nitrogen starvation response of Bacillus licheniformis. Proteomics 2007, 7, 413–423. [Google Scholar] [CrossRef] [PubMed]
- Song, P.; Chen, C.; Tian, Q.; Lin, M.; Huang, H.; Li, S. Two-stage oxygen supply strategy for enhanced lipase production by Bacillus subtilis based on metabolic flux analysis. Biochem. Eng. J. 2013, 71, 1–10. [Google Scholar] [CrossRef]
- Diomandã, S.; Nguyen-The, C.; Guinebretiã, M.H.; Broussolle, V.; Brillard, J. Role of fatty acids in Bacillus environmental adaptation. Front. Microbiol. 2015, 6, 813. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.-Y.; Kim, Y.-S.; Shin, D.-H. Antimicrobial Synergistic Effect of Linolenic Acid and Monoglyceride against Bacillus cereus and Staphylococcus aureus. J. Agric. Food Chem. 2002, 50, 2193–2199. [Google Scholar] [CrossRef]
- Vamanu, E.; Gatea, F. Correlations between Microbiota Bioactivity and Bioavailability of Functional Compounds: A Mini-Review. Biomedicines 2020, 8, 39. [Google Scholar] [CrossRef] [Green Version]
- McEwan, C.E.A.; Gatherer, D.; McEwan, N.R. Nitrogen-fixing aerobic bacteria have higher genomic GC content than non-fixing species within the same genus. Hereditas 2004, 128, 173–178. [Google Scholar] [CrossRef] [Green Version]
- Rocha, E.P.; Danchin, A. Base composition bias might result from competition for metabolic resources. Trends Genet. 2002, 18, 291–294. [Google Scholar] [CrossRef]
- Woolfit, M.; Bromham, L. Increased Rates of Sequence Evolution in Endosymbiotic Bacteria and Fungi with Small Effective Population Sizes. Mol. Biol. Evol. 2003, 20, 1545–1555. [Google Scholar] [CrossRef] [PubMed]
- Bentley, S.D.; Parkhill, J. Comparative Genomic Structure of Prokaryotes. Annu. Rev. Genet. 2004, 38, 771–791. [Google Scholar] [CrossRef] [Green Version]
- Foerstner, K.U.; Von Mering, C.; Hooper, S.D.; Bork, P. Environments shape the nucleotide composition of genomes. EMBO Rep. 2005, 6, 1208–1213. [Google Scholar] [CrossRef]
- Musto, H.; Naya, H.; Zavala, A.; Romero, H.; Alvarez-Valin, F.; Bernardi, G. Genomic GC level, optimal growth temperature, and genome size in prokaryotes. Biochem. Biophys. Res. Commun. 2006, 347, 1. [Google Scholar] [CrossRef] [PubMed]
- Tettelin, H.; Riley, D.; Cattuto, C.; Medini, D. Comparative genomics: The bacterial pan-genome. Curr. Opin. Microbiol. 2008, 11, 472–477. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Metabolic Pathway | Number of Proteins | |
|---|---|---|
| Cytosol | Membrane | |
| 00010 Glycolysis/Gluconeogenesis | 8 | 14 |
| 00020 Citrate cycle (TCA cycle) | 3 | 11 |
| 00030 Pentose phosphate pathway | 3 | 7 |
| 00051 Fructose and mannose metabolism | 2 | 3 |
| 00052 Galactose metabolism | 0 | 2 |
| 00061 Fatty acid biosynthesis | 1 | 2 |
| 00071 Fatty acid degradation | 1 | 1 |
| 00190 Oxidative phosphorylation | 3 | 5 |
| 00220 Arginine biosynthesis | 1 | 1 |
| 00261 Monobactam biosynthesis | 1 | 1 |
| 00290 Valine, leucine, and isoleucine biosynthesis | 1 | 2 |
| 00500 Starch and sucrose metabolism | 0 | 3 |
| 00561 Glycerolipid metabolism | 3 | 4 |
| 00564 Glycerophospholipid metabolism | 1 | 2 |
| 00630 Glyoxylate and dicarboxylate metabolism | 2 | 5 |
| 00790 Folate biosynthesis | 1 | 1 |
| 00910 Nitrogen metabolism | 1 | 1 |
| 00920 Sulfur metabolism | 1 | 1 |
| 00983 Drug metabolism—other enzymes | 2 | 2 |
| 01051 Biosynthesis of ansamycins | 0 | 1 |
| 01501 beta-Lactam resistance | 1 | 1 |
| 02010 ABC transporters | 2 | 3 |
| 02024 Quorum sensing | 3 | 6 |
| 02040 Flagellar assembly | 2 | 2 |
| 02060 Phosphotransferase system (PTS) | 2 | 3 |
| Function | No. of Genes | % | No. of Cytosol Proteins | % | No. of Membrane Proteins | % |
|---|---|---|---|---|---|---|
| Chromatin structure and dynamics | 1 | 0.03 | ||||
| Energy production and conversion | 159 | 5.06 | 11 | 9.09 | 29 | 10.36 |
| Cell cycle control, cell division, chromosome partitioning | 52 | 1.65 | 2 | 1.65 | 4 | 1.43 |
| Amino acid transport and metabolism | 308 | 9.80 | 6 | 4.96 | 19 | 6.79 |
| Nucleotide transport and metabolism | 85 | 2.70 | 9 | 7.44 | 15 | 5.36 |
| Carbohydrate transport and metabolism | 249 | 7.92 | 14 | 11.57 | 27 | 9.64 |
| Coenzyme transport and metabolism | 170 | 5.41 | 5 | 4.13 | 9 | 3.21 |
| Lipid transport and metabolism | 121 | 3.85 | 1 | 0.83 | 5 | 1.79 |
| Translation, ribosomal structure, and biogenesis | 228 | 7.25 | 30 | 24.79 | 56 | 20.00 |
| Transcription | 279 | 8.87 | 11 | 9.09 | 24 | 8.57 |
| Replication, recombination, and repair | 110 | 3.50 | 6 | 4.96 | 18 | 6.43 |
| Cell wall/membrane/envelope biogenesis | 181 | 5.76 | 1 | 0.83 | 4 | 1.43 |
| Cell motility | 61 | 1.94 | 0 | 0.00 | 0 | 0.00 |
| Posttranslational modification, protein turnover, chaperones | 118 | 3.75 | 11 | 9.09 | 16 | 5.71 |
| Inorganic ion transport and metabolism | 158 | 5.03 | 3 | 2.48 | 9 | 3.21 |
| Secondary metabolites biosynthesis, transport, and catabolism | 73 | 2.32 | 2 | 1.65 | 5 | 1.79 |
| General function prediction only | 277 | 8.81 | 1 | 0.83 | 10 | 3.57 |
| Function unknown | 183 | 5.82 | 2 | 1.65 | 5 | 1.79 |
| Signal transduction mechanisms | 177 | 5.63 | 4 | 3.31 | 15 | 5.36 |
| Intracellular trafficking, secretion, and vesicular transport | 27 | 0.86 | 1 | 0.83 | 2 | 0.71 |
| Defense mechanisms | 69 | 2.19 | 1 | 0.83 | 6 | 2.14 |
| Extracellular structures | 2 | 0.06 | 0 | 0.00 | 0 | 0.00 |
| Mobilome: Prophages, transposons | 56 | 1.78 | 0 | 0.00 | 2 | 0.71 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Reyes-Cortes, J.L.; Azaola-Espinosa, A.; Lozano-Aguirre, L.; Ponce-Alquicira, E. Physiological and Genomic Analysis of Bacillus pumilus UAMX Isolated from the Gastrointestinal Tract of Overweight Individuals. Microorganisms 2021, 9, 1076. https://doi.org/10.3390/microorganisms9051076
Reyes-Cortes JL, Azaola-Espinosa A, Lozano-Aguirre L, Ponce-Alquicira E. Physiological and Genomic Analysis of Bacillus pumilus UAMX Isolated from the Gastrointestinal Tract of Overweight Individuals. Microorganisms. 2021; 9(5):1076. https://doi.org/10.3390/microorganisms9051076
Chicago/Turabian StyleReyes-Cortes, José Luis, Alejandro Azaola-Espinosa, Luis Lozano-Aguirre, and Edith Ponce-Alquicira. 2021. "Physiological and Genomic Analysis of Bacillus pumilus UAMX Isolated from the Gastrointestinal Tract of Overweight Individuals" Microorganisms 9, no. 5: 1076. https://doi.org/10.3390/microorganisms9051076