
Effects of Visceralising Leishmania on the Spleen, Liver, and Bone Marrow: A Pathophysiological Perspective


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Immunobiology of Hepatic versus Splenic Leishmanial Responses
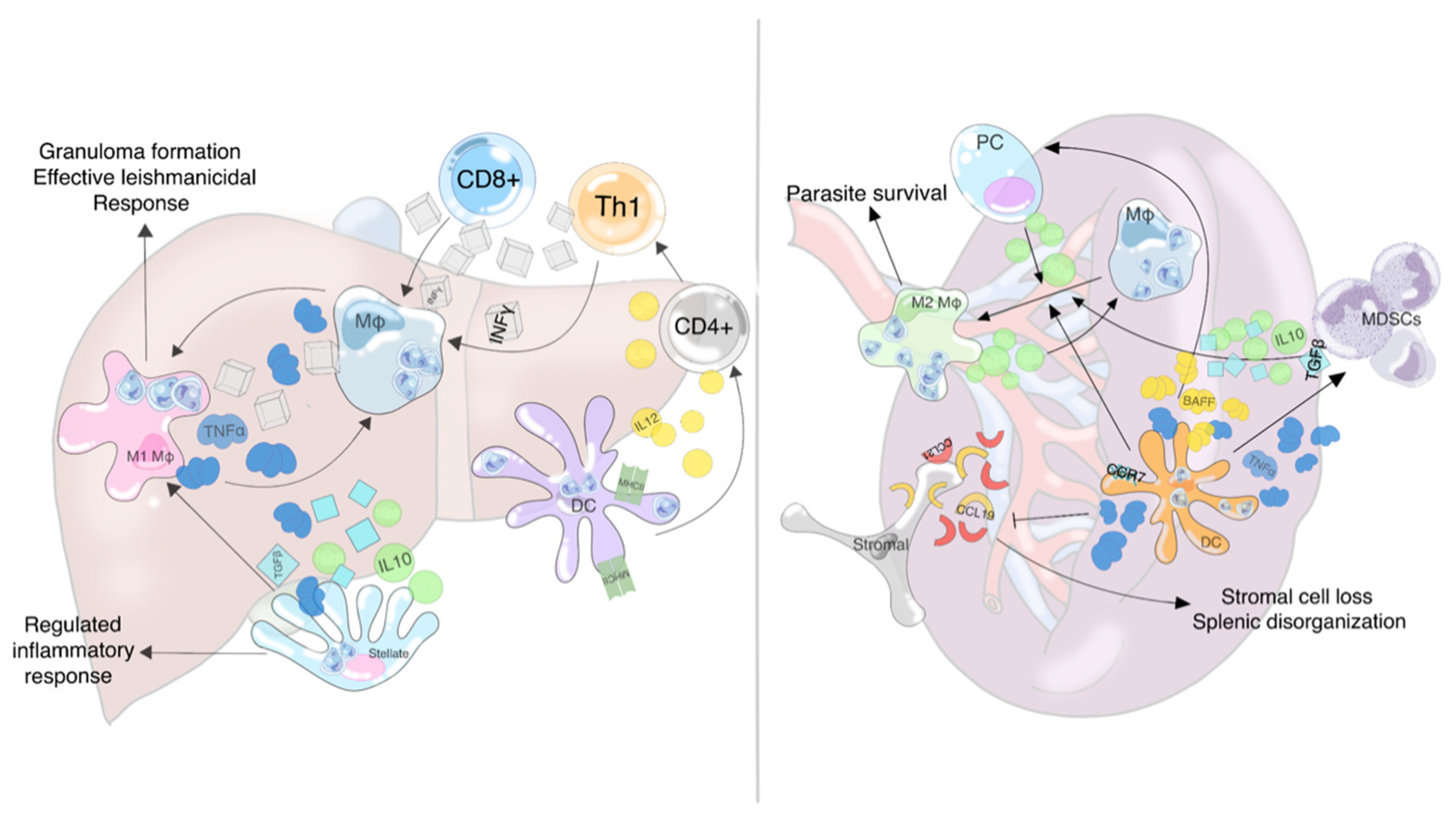
2.1. Overview of the Leishmanicidal Response in the Liver
2.2. Overview of the Ineffective Splenic Response
3. Pathophysiology of Pancytopenia in VL
3.1. Pathophysiology of Thrombocytopenia
3.2. Hemichromes and the Effects of Increased Oxidative Stress on the Erythroid Lineage
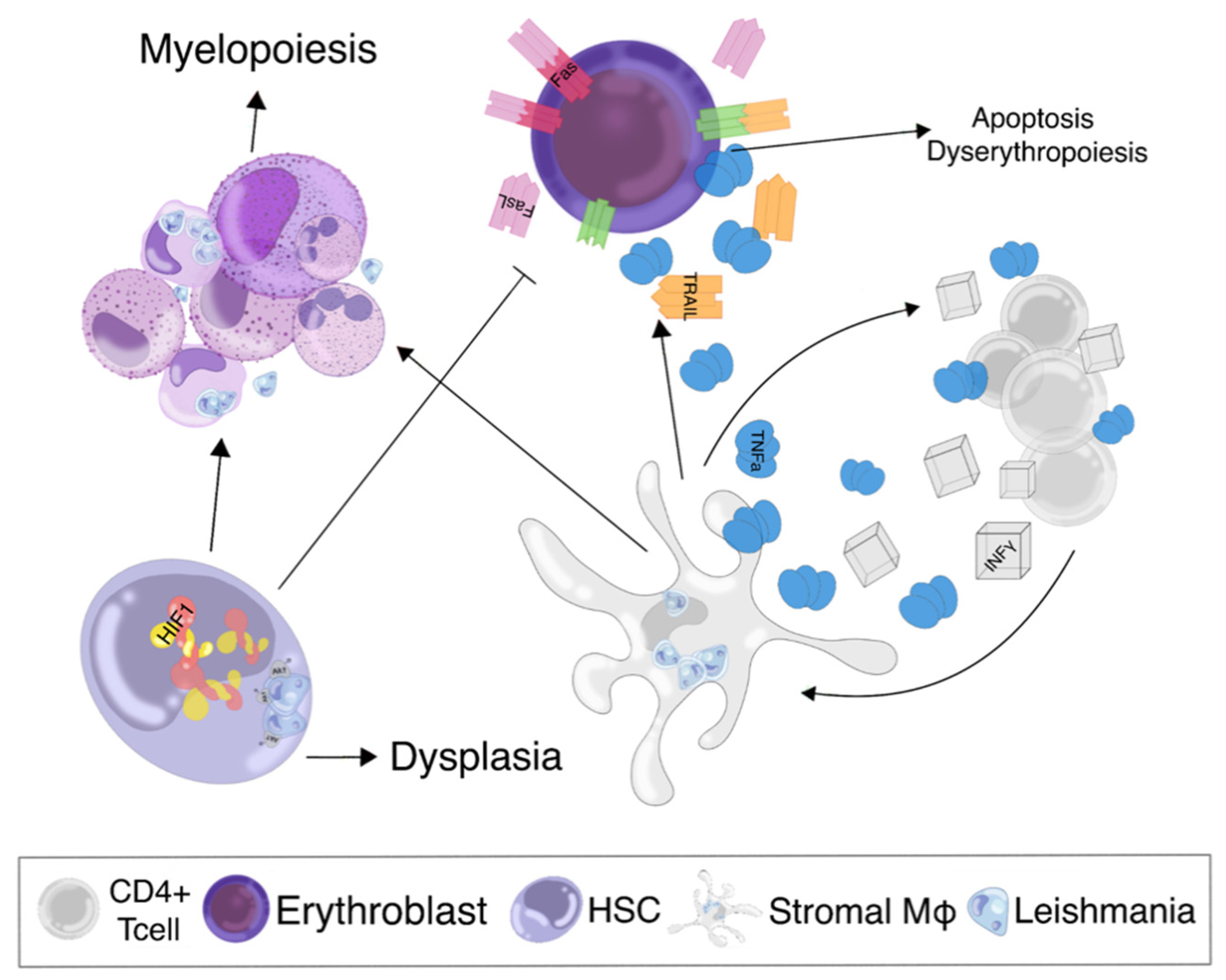
3.3. Bone Marrow Suppression in Visceral Leishmaniasis
3.4. Bone Marrow Dysplasia in Visceral Leishmaniasis
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- WHO. The disease and its epidemiology. Available online: https://www.who.int/leishmaniasis/disease_epidemiology/en/ (accessed on 16 January 2021).
- Akuffo, H.; Costa, C.; van Griensven, J.; Burza, S.; Moreno, J.; Herrero, M. New insights into leishmaniasis in the immunosuppressed. PLoS Negl. Trop. Dis. 2018, 12, e0006375. [Google Scholar] [CrossRef]
- World Health Organization. Leishmaniasis. Available online: https://www.who.int/news-room/fact-sheets/detail/leishmaniasis (accessed on 16 January 2021).
- Pigott, D.M.; Bhatt, S.; Golding, N.; Duda, K.A.; Battle, K.E.; Brady, O.J.; Messina, J.P.; Balard, Y.; Bastien, P.; Pratlong, F.; et al. Global distribution maps of the leishmaniases. Elife 2014, 3, e02851. [Google Scholar] [CrossRef] [PubMed]
- Singh, O.P.; Hasker, E.; Sacks, D.; Boelaert, M.; Sundar, S. Asymptomatic Leishmania infection: A new challenge for Leishmania control. Clin. Infect. Dis. 2014, 58, 1424–1429. [Google Scholar] [CrossRef]
- Bankoti, R.; Stager, S. Differential Regulation of the Immune Response in the Spleen and Liver of Mice Infected with Leishmania donovani. J. Trop. Med. 2012, 2012, 639304. [Google Scholar] [CrossRef] [Green Version]
- Engwerda, C.R.; Ato, M.; Kaye, P.M. Macrophages, pathology and parasite persistence in experimental visceral leishmaniasis. Trends Parasitol. 2004, 20, 524–530. [Google Scholar] [CrossRef] [PubMed]
- Kaye, P.M.; Svensson, M.; Ato, M.; Maroof, A.; Polley, R.; Stager, S.; Zubairi, S.; Engwerda, C.R. The immunopathology of experimental visceral leishmaniasis. Immunol. Rev. 2004, 201, 239–253. [Google Scholar] [CrossRef] [PubMed]
- Sundar, S.; Singh, B. Emerging therapeutic targets for treatment of leishmaniasis. Expert Opin. Ther. Targets 2018, 22, 467–486. [Google Scholar] [CrossRef] [PubMed]
- Moreira, M.L.; Costa-Pereira, C.; Alves, M.L.; Marteleto, B.H.; Ribeiro, V.M.; Peruhype-Magalhaes, V.; Giunchetti, R.C.; Martins-Filho, O.A.; Araujo, M.S. Vaccination against canine leishmaniosis increases the phagocytic activity, nitric oxide production and expression of cell activation/migration molecules in neutrophils and monocytes. Vet. Parasitol. 2016, 220, 33–45. [Google Scholar] [CrossRef]
- Lypaczewski, P.; Hoshizaki, J.; Zhang, W.W.; McCall, L.I.; Torcivia-Rodriguez, J.; Simonyan, V.; Kaur, A.; Dewar, K.; Matlashewski, G. A complete Leishmania donovani reference genome identifies novel genetic variations associated with virulence. Sci. Rep. 2018, 8, 16549. [Google Scholar] [CrossRef]
- Zhang, W.W.; Matlashewski, G. Characterization of the A2-A2rel gene cluster in Leishmania donovani: Involvement of A2 in visceralization during infection. Mol. Microbiol. 2001, 39, 935–948. [Google Scholar] [CrossRef]
- Santana, C.C.; Vassallo, J.; de Freitas, L.A.; Oliveira, G.G.; Pontes-de-Carvalho, L.C.; dos-Santos, W.L. Inflammation and structural changes of splenic lymphoid tissue in visceral leishmaniasis: A study on naturally infected dogs. Parasite Immunol. 2008, 30, 515–524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silva-O’Hare, J.; de Oliveira, I.S.; Klevorn, T.; Almeida, V.A.; Oliveira, G.G.; Atta, A.M.; de Freitas, L.A.; Dos-Santos, W.L. Disruption of Splenic Lymphoid Tissue and Plasmacytosis in Canine Visceral Leishmaniasis: Changes in Homing and Survival of Plasma Cells. PLoS ONE 2016, 11, e0156733. [Google Scholar] [CrossRef] [PubMed]
- Cavalcanti, A.S.; Ribeiro-Alves, M.; Pereira, L.d.O.; Mestre, G.L.; Ferreira, A.B.; Morgado, F.N.; Boite, M.C.; Cupolillo, E.; Moraes, M.O.; Porrozzi, R. Parasite load induces progressive spleen architecture breakage and impairs cytokine mRNA expression in Leishmania infantum-naturally infected dogs. PLoS ONE 2015, 10, e0123009. [Google Scholar] [CrossRef] [PubMed]
- Hermida, M.D.; de Melo, C.V.B.; Lima, I.D.S.; Oliveira, G.G.S.; Dos-Santos, W.L.C. Histological Disorganization of Spleen Compartments and Severe Visceral Leishmaniasis. Front. Cell. Infect. Microbiol. 2018, 8, 394. [Google Scholar] [CrossRef] [Green Version]
- Koster, K.L.; Laws, H.J.; Troeger, A.; Meisel, R.; Borkhardt, A.; Oommen, P.T. Visceral Leishmaniasis as a Possible Reason for Pancytopenia. Front. Pediatr. 2015, 3, 59. [Google Scholar] [CrossRef] [Green Version]
- Varma, N.; Naseem, S. Hematologic changes in visceral leishmaniasis/kala azar. Indian J. Hematol. Blood Transfus. 2010, 26, 78–82. [Google Scholar] [CrossRef] [Green Version]
- Pippard, M.J.; Moir, D.; Weatherall, D.J.; Lenicker, H.M. Mechanism of anaemia in resistant visceral leishmaniasis. Ann. Trop. Med. Parasitol. 1986, 80, 317–323. [Google Scholar] [CrossRef]
- Goto, Y.; Cheng, J.; Omachi, S.; Morimoto, A. Prevalence, severity, and pathogeneses of anemia in visceral leishmaniasis. Parasitol. Res. 2017, 116, 457–464. [Google Scholar] [CrossRef] [PubMed]
- Yarali, N.; Fisgin, T.; Duru, F.; Kara, A. Myelodysplastic features in visceral leishmaniasis. Am. J. Hematol. 2002, 71, 191–195. [Google Scholar] [CrossRef]
- Sheikha, A. Dyserythropoiesis in 105 patients with visceral leishmaniasis. Lab. Hematol. 2004, 10, 206–211. [Google Scholar]
- Preham, O.; Pinho, F.A.; Pinto, A.I.; Rani, G.F.; Brown, N.; Hitchcock, I.S.; Goto, H.; Kaye, P.M. CD4(+) T Cells Alter the Stromal Microenvironment and Repress Medullary Erythropoiesis in Murine Visceral Leishmaniasis. Front. Immunol. 2018, 9, 2958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murray, H.W. Tissue granuloma structure-function in experimental visceral leishmaniasis. Int. J. Exp. Pathol. 2001, 82, 249–267. [Google Scholar] [CrossRef]
- Salguero, F.J.; Garcia-Jimenez, W.L.; Lima, I.; Seifert, K. Histopathological and immunohistochemical characterisation of hepatic granulomas in Leishmania donovani-infected BALB/c mice: A time-course study. Parasites Vectors 2018, 11, 73. [Google Scholar] [CrossRef] [PubMed]
- Kaye, P.M.; Beattie, L. Lessons from other diseases: Granulomatous inflammation in leishmaniasis. Semin. Immunopathol. 2016, 38, 249–260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bunn, P.T.; Stanley, A.C.; de Labastida Rivera, F.; Mulherin, A.; Sheel, M.; Alexander, C.E.; Faleiro, R.J.; Amante, F.H.; Montes De Oca, M.; Best, S.E.; et al. Tissue requirements for establishing long-term CD4+ T cell-mediated immunity following Leishmania donovani infection. J. Immunol. 2014, 192, 3709–3718. [Google Scholar] [CrossRef] [PubMed]
- Robert-Gangneux, F.; Drogoul, A.S.; Rostan, O.; Piquet-Pellorce, C.; Cayon, J.; Lisbonne, M.; Herbelin, A.; Gascan, H.; Guiguen, C.; Samson, M.; et al. Invariant NKT cells drive hepatic cytokinic microenvironment favoring efficient granuloma formation and early control of Leishmania donovani infection. PLoS ONE 2012, 7, e33413. [Google Scholar] [CrossRef]
- Moore, J.W.; Moyo, D.; Beattie, L.; Andrews, P.S.; Timmis, J.; Kaye, P.M. Functional complexity of the Leishmania granuloma and the potential of in silico modeling. Front. Immunol. 2013, 4, 35. [Google Scholar] [CrossRef] [Green Version]
- Krovi, S.H.; Gapin, L. Invariant Natural Killer T Cell Subsets—More Than Just Developmental Intermediates. Front. Immunol. 2018, 9, 1393. [Google Scholar] [CrossRef] [Green Version]
- Stanley, A.C.; Zhou, Y.; Amante, F.H.; Randall, L.M.; Haque, A.; Pellicci, D.G.; Hill, G.R.; Smyth, M.J.; Godfrey, D.I.; Engwerda, C.R. Activation of invariant NKT cells exacerbates experimental visceral leishmaniasis. PLoS Pathog. 2008, 4, e1000028. [Google Scholar] [CrossRef] [Green Version]
- Iyoda, T.; Ushida, M.; Kimura, Y.; Minamino, K.; Hayuka, A.; Yokohata, S.; Ehara, H.; Inaba, K. Invariant NKT cell anergy is induced by a strong TCR-mediated signal plus co-stimulation. Int. Immunol. 2010, 22, 905–913. [Google Scholar] [CrossRef]
- Tiburcio, R.; Nunes, S.; Nunes, I.; Ampuero, M.R.; Silva, I.B.; Lima, R.; Tavares, N.M.; Brodskyn, C. Molecular Aspects of Dendritic Cell Activation in Leishmaniasis: An Immunobiological View. Front. Immunol. 2019, 10, 227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukherjee, P.; Ghosh, A.K.; Ghose, A.C. Infection pattern and immune response in the spleen and liver of BALB/c mice intracardially infected with Leishmania donovani amastigotes. Immunol. Lett. 2003, 86, 131–138. [Google Scholar] [CrossRef]
- Khadem, F.; Gao, X.; Mou, Z.; Jia, P.; Movassagh, H.; Onyilagha, C.; Gounni, A.S.; Wright, M.C.; Uzonna, J.E. Hepatic stellate cells regulate liver immunity to visceral leishmaniasis through P110delta-dependent induction and expansion of regulatory T cells in mice. Hepatology 2016, 63, 620–632. [Google Scholar] [CrossRef] [PubMed]
- El Hag, I.A.; Hashim, F.A.; El Toum, I.A.; Homeida, M.; El Kalifa, M.; El Hassan, A.M. Liver morphology and function in visceral leishmaniasis (Kala-azar). J. Clin. Pathol. 1994, 47, 547–551. [Google Scholar] [CrossRef] [Green Version]
- Terrazas, C.; Varikuti, S.; Oghumu, S.; Steinkamp, H.M.; Ardic, N.; Kimble, J.; Nakhasi, H.; Satoskar, A.R. Ly6C(hi) inflammatory monocytes promote susceptibility to Leishmania donovani infection. Sci. Rep. 2017, 7, 14693. [Google Scholar] [CrossRef] [Green Version]
- Kima, P.E. PI3K signaling in Leishmania infections. Cell. Immunol. 2016, 309, 19–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McMahon-Pratt, D.; Alexander, J. Does the Leishmania major paradigm of pathogenesis and protection hold for New World cutaneous leishmaniases or the visceral disease? Immunol. Rev. 2004, 201, 206–224. [Google Scholar] [CrossRef]
- Paun, A.; Bankoti, R.; Joshi, T.; Pitha, P.M.; Stager, S. Critical role of IRF-5 in the development of T helper 1 responses to Leishmania donovani infection. PLoS Pathog. 2011, 7, e1001246. [Google Scholar] [CrossRef] [Green Version]
- Montes de Oca, M.; Engwerda, C.R.; Kaye, P.M. Cytokines and splenic remodelling during Leishmania donovani infection. Cytokine X 2020, 2, 100036. [Google Scholar] [CrossRef]
- Singh, A.K.; Mukhopadhyay, C.; Biswas, S.; Singh, V.K.; Mukhopadhyay, C.K. Intracellular pathogen Leishmania donovani activates hypoxia inducible factor-1 by dual mechanism for survival advantage within macrophage. PLoS ONE 2012, 7, e38489. [Google Scholar] [CrossRef]
- Corzo, C.A.; Condamine, T.; Lu, L.; Cotter, M.J.; Youn, J.I.; Cheng, P.; Cho, H.I.; Celis, E.; Quiceno, D.G.; Padhya, T.; et al. HIF-1alpha regulates function and differentiation of myeloid-derived suppressor cells in the tumor microenvironment. J. Exp. Med. 2010, 207, 2439–2453. [Google Scholar] [CrossRef] [PubMed]
- Schatz, V.; Neubert, P.; Rieger, F.; Jantsch, J. Hypoxia, Hypoxia-Inducible Factor-1alpha, and Innate Antileishmanial Immune Responses. Front. Immunol. 2018, 9, 216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prina, E.; Abdi, S.Z.; Lebastard, M.; Perret, E.; Winter, N.; Antoine, J.C. Dendritic cells as host cells for the promastigote and amastigote stages of Leishmania amazonensis: The role of opsonins in parasite uptake and dendritic cell maturation. J. Cell Sci. 2004, 117 Pt 2, 315–325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Backer, R.A.; Diener, N.; Clausen, B.E. Langerin(+)CD8(+) Dendritic Cells in the Splenic Marginal Zone: Not So Marginal After All. Front. Immunol. 2019, 10, 741. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Nylen, S. Immunobiology of visceral leishmaniasis. Front. Immunol. 2012, 3, 251. [Google Scholar] [CrossRef] [Green Version]
- Pillarisetty, V.G.; Shah, A.B.; Miller, G.; Bleier, J.I.; DeMatteo, R.P. Liver dendritic cells are less immunogenic than spleen dendritic cells because of differences in subtype composition. J. Immunol. 2004, 172, 1009–1017. [Google Scholar] [CrossRef] [PubMed]
- Abidin, B.M.; Hammami, A.; Stager, S.; Heinonen, K.M. Infection-adapted emergency hematopoiesis promotes visceral leishmaniasis. PLoS Pathog. 2017, 13, e1006422. [Google Scholar] [CrossRef]
- Mai, L.T.; Smans, M.; Silva-Barrios, S.; Fabié, A.; Stäger, S. IRF-5 Expression in Myeloid Cells Is Required for Splenomegaly in L. donovani Infected Mice. Front. Immunol. 2020, 10, 10. [Google Scholar] [CrossRef] [Green Version]
- Ato, M.; Maroof, A.; Zubairi, S.; Nakano, H.; Kakiuchi, T.; Kaye, P.M. Loss of dendritic cell migration and impaired resistance to Leishmania donovani infection in mice deficient in CCL19 and CCL21. J. Immunol. 2006, 176, 5486–5493. [Google Scholar] [CrossRef] [Green Version]
- Osorio, E.Y.; Medina-Colorado, A.A.; Travi, B.L.; Melby, P.C. In-situ proliferation contributes to the accumulation of myeloid cells in the spleen during progressive experimental visceral leishmaniasis. PLoS ONE 2020, 15, e0242337. [Google Scholar] [CrossRef]
- Kong, F.; Saldarriaga, O.A.; Spratt, H.; Osorio, E.Y.; Travi, B.L.; Luxon, B.A.; Melby, P.C. Transcriptional Profiling in Experimental Visceral Leishmaniasis Reveals a Broad Splenic Inflammatory Environment that Conditions Macrophages toward a Disease-Promoting Phenotype. PLoS Pathog. 2017, 13, e1006165. [Google Scholar] [CrossRef]
- Gantt, K.R.; Schultz-Cherry, S.; Rodriguez, N.; Jeronimo, S.M.; Nascimento, E.T.; Goldman, T.L.; Recker, T.J.; Miller, M.A.; Wilson, M.E. Activation of TGF-beta by Leishmania chagasi: Importance for parasite survival in macrophages. J. Immunol. 2003, 170, 2613–2620. [Google Scholar] [CrossRef] [Green Version]
- Das, V.N.R.; Bimal, S.; Siddiqui, N.A.; Kumar, A.; Pandey, K.; Sinha, S.K.; Topno, R.K.; Mahentesh, V.; Singh, A.K.; Lal, C.S.; et al. Conversion of asymptomatic infection to symptomatic visceral leishmaniasis: A study of possible immunological markers. PLoS Negl. Trop. Dis. 2020, 14, e0008272. [Google Scholar] [CrossRef]
- Hammami, A.; Abidin, B.M.; Charpentier, T.; Fabie, A.; Duguay, A.P.; Heinonen, K.M.; Stager, S. HIF-1alpha is a key regulator in potentiating suppressor activity and limiting the microbicidal capacity of MDSC-like cells during visceral leishmaniasis. PLoS Pathog. 2017, 13, e1006616. [Google Scholar] [CrossRef] [Green Version]
- Harris, A.J.; Thompson, A.R.; Whyte, M.K.; Walmsley, S.R. HIF-mediated innate immune responses: Cell signaling and therapeutic implications. Hypoxia 2014, 2, 47–58. [Google Scholar]
- Basu, A.; Chakrabarti, G.; Saha, A.; Bandyopadhyay, S. Modulation of CD11C+ splenic dendritic cell functions in murine visceral leishmaniasis: Correlation with parasite replication in the spleen. Immunology 2000, 99, 305–313. [Google Scholar] [CrossRef] [PubMed]
- Hammami, A.; Abidin, B.M.; Heinonen, K.M.; Stager, S. HIF-1alpha hampers dendritic cell function and Th1 generation during chronic visceral leishmaniasis. Sci. Rep. 2018, 8, 3500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vogt, P.K.; Hart, J.R. PI3K and STAT3: A new alliance. Cancer Discov. 2011, 1, 481–486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, A.; Das, S.; Mandal, A.; Verma, S.; Abhishek, K.; Kumar, A.; Kumar, V.; Ghosh, A.K.; Das, P. Leishmania infection activates host mTOR for its survival by M2 macrophage polarization. Parasite Immunol. 2018, 40, e12586. [Google Scholar] [CrossRef]
- Nandan, D.; Camargo de Oliveira, C.; Moeenrezakhanlou, A.; Lopez, M.; Silverman, J.M.; Subek, J.; Reiner, N.E. Myeloid cell IL-10 production in response to leishmania involves inactivation of glycogen synthase kinase-3beta downstream of phosphatidylinositol-3 kinase. J. Immunol. 2012, 188, 367–378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beattie, L.; d’El-Rei Hermida, M.; Moore, J.W.; Maroof, A.; Brown, N.; Lagos, D.; Kaye, P.M. A transcriptomic network identified in uninfected macrophages responding to inflammation controls intracellular pathogen survival. Cell Host Microbe 2013, 14, 357–368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, H.; Liu, W.; Su, Y.; Wei, Z.; Liu, J.; Kolluri, S.K.; Wu, H.; Cao, Y.; Chen, J.; Wu, Y.; et al. NSAID sulindac and its analog bind RXRalpha and inhibit RXRalpha-dependent AKT signaling. Cancer Cell 2010, 17, 560–573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khadem, F.; Jia, P.; Mou, Z.; Feiz Barazandeh, A.; Liu, D.; Keynan, Y.; Uzonna, J.E. Pharmacological inhibition of p110delta subunit of PI3K confers protection against experimental leishmaniasis. J. Antimicrob. Chemother. 2017, 72, 467–477. [Google Scholar] [CrossRef] [Green Version]
- Martinez de Narvajas, I.; Diaz, A.; Bassegoda, O.; Carpio, A.; Fuster, C.; Valls, M.E.; Alvarez-Martinez, M.J.; Garcia-Vidal, C.; Soriano, A.; Martinez, J.A.; et al. Acute liver failure due to visceral leishmaniasis in Barcelona: A case report. BMC Infect. Dis. 2019, 19, 874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathur, P.; Samantaray, J.C.; Samanta, P. High prevalence of functional liver derangement in visceral leishmaniasis at an Indian tertiary care center. Clin. Gastroenterol. Hepatol. 2008, 6, 1170–1172. [Google Scholar] [CrossRef]
- Gupta, R.; Musallam, K.M.; Taher, A.T.; Rivella, S. Ineffective Erythropoiesis: Anemia and Iron Overload. Hematol. Oncol. Clin. N. Am. 2018, 32, 213–221. [Google Scholar] [CrossRef] [PubMed]
- Rani, G.F.; Preham, O.; Hitchcock, I.; Kaye, P. Understanding the Mechanisms Underlying Thrombocytopenia in Visceral Leishmaniasis. Blood 2019, 134 (Suppl. 1), 2378. [Google Scholar] [CrossRef]
- Beaulieu, L.M.; Freedman, J.E. The role of inflammation in regulating platelet production and function: Toll-like receptors in platelets and megakaryocytes. Thromb. Res. 2010, 125, 205–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saeed, A.M.; Khalil, E.A.; Elhassan, A.M.; Hashim, F.A.; Elhassan, A.M.; Fandrey, J.; Jelkmann, W. Serum erythropoietin concentration in anaemia of visceral leishmaniasis (kala-azar) before and during antimonial therapy. Br. J. Haematol. 1998, 100, 720–724. [Google Scholar] [CrossRef] [PubMed]
- Ginzburg, Y.; Rivella, S. beta-thalassemia: A model for elucidating the dynamic regulation of ineffective erythropoiesis and iron metabolism. Blood 2011, 118, 4321–4330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rachmilewitz, E.A.; Thorell, B. Hemichromes in single inclusion bodies in red cells of beta thalassemia. Blood 1972, 39, 794–800. [Google Scholar] [CrossRef] [PubMed]
- Cappellini, M.D.; Tavazzi, D.; Duca, L.; Graziadei, G.; Mannu, F.; Turrini, F.; Arese, P.; Fiorelli, G. Metabolic indicators of oxidative stress correlate with haemichrome attachment to membrane, band 3 aggregation and erythrophagocytosis in beta-thalassaemia intermedia. Br. J. Haematol. 1999, 104, 504–512. [Google Scholar] [CrossRef] [PubMed]
- Saha Roy, S.; Chowdhury, K.D.; Sen, G.; Biswas, T. Oxidation of hemoglobin and redistribution of band 3 promote erythrophagocytosis in visceral leishmaniasis. Mol. Cell. Biochem. 2009, 321, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Low, P.S. Role of hemoglobin denaturation and band 3 clustering in initiating red cell removal. Adv. Exp. Med. Biol. 1991, 307, 173–183. [Google Scholar] [PubMed]
- Pantaleo, A.; Ferru, E.; Pau, M.C.; Khadjavi, A.; Mandili, G.; Matte, A.; Spano, A.; De Franceschi, L.; Pippia, P.; Turrini, F. Band 3 Erythrocyte Membrane Protein Acts as Redox Stress Sensor Leading to Its Phosphorylation by p (72) Syk. Oxidative Med. Cell. Longev. 2016, 2016, 6051093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cotterell, S.E.J.; Engwerda, C.R.; Kaye, P.M. Leishmania donovani infection of bone marrow stromal macrophages selectively enhances myelopoiesis, by a mechanism involving GM-CSF and TNF-α. Blood 2000, 95, 1642–1651. [Google Scholar] [CrossRef] [PubMed]
- Lafuse, W.P.; Story, R.; Mahylis, J.; Gupta, G.; Varikuti, S.; Steinkamp, H.; Oghumu, S.; Satoskar, A.R. Leishmania donovani infection induces anemia in hamsters by differentially altering erythropoiesis in bone marrow and spleen. PLoS ONE 2013, 8, e59509. [Google Scholar] [CrossRef] [Green Version]
- Carvalho-Gontijo, R.; Moreira, D.R.; Resende, M.; Costa-Silva, M.F.; Peruhype-Magalhaes, V.; Ribeiro, C.M.F.; Ribeiro, D.D.; Silvestre, R.; Cordeiro-da-Silva, A.; Martins-Filho, O.A.; et al. Infection of hematopoietic stem cells by Leishmania infantum increases erythropoiesis and alters the phenotypic and functional profiles of progeny. Cell. Immunol. 2018, 326, 77–85. [Google Scholar] [CrossRef] [Green Version]
- Iommarini, L.; Porcelli, A.M.; Gasparre, G.; Kurelac, I. Non-Canonical Mechanisms Regulating Hypoxia-Inducible Factor 1 Alpha in Cancer. Front. Oncol. 2017, 7, 286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costa, T.L.; Ribeiro-Dias, F.; Oliveira, M.A.; Bezerra, J.C.; Vinaud, M.C. Energetic metabolism of axenic promastigotes of Leishmania (Viannia) braziliensis. Exp. Parasitol. 2011, 128, 438–443. [Google Scholar] [CrossRef] [Green Version]
- Bringaud, F.; Riviere, L.; Coustou, V. Energy metabolism of trypanosomatids: Adaptation to available carbon sources. Mol. Biochem. Parasitol. 2006, 149, 1–9. [Google Scholar] [CrossRef]
- Subramanian, A.; Jhawar, J.; Sarkar, R.R. Dissecting Leishmania infantum Energy Metabolism—A Systems Perspective. PLoS ONE 2015, 10, e0137976. [Google Scholar]
- Michels, P.A.; Bringaud, F.; Herman, M.; Hannaert, V. Metabolic functions of glycosomes in trypanosomatids. Biochim. Biophys. Acta 2006, 1763, 1463–1477. [Google Scholar] [CrossRef] [PubMed]
- Gabaldon, T.; Pittis, A.A. Origin and evolution of metabolic sub-cellular compartmentalization in eukaryotes. Biochimie 2015, 119, 262–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mehlhorn, H. Cellular Organization of Parasitic Protozoa. In Topley & Wilson’s Microbiology and Microbial Infections, 1st ed.; Mahy, B.W., Ter Meulen, V., Borriello, S.P., Murray, P.R., Funke, G., Kaufmann, S.H., Steward, M.W., Merz, W.G., Hay, R.J., Cox, F., Eds.; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2010; Volume 6. [Google Scholar]
- Mesias, A.C.; Garg, N.J.; Zago, M.P. Redox Balance Keepers and Possible Cell Functions Managed by Redox Homeostasis in Trypanosoma cruzi. Front. Cell. Infect. Microbiol. 2019, 9, 435. [Google Scholar] [CrossRef] [PubMed]
- McConville, M.J.; Naderer, T. Metabolic pathways required for the intracellular survival of Leishmania. Annu. Rev. Microbiol. 2011, 65, 543–561. [Google Scholar] [CrossRef]
- Saunders, E.C.; Ng, W.W.; Kloehn, J.; Chambers, J.M.; Ng, M.; McConville, M.J. Induction of a stringent metabolic response in intracellular stages of Leishmania mexicana leads to increased dependence on mitochondrial metabolism. PLoS Pathog. 2014, 10, e1003888. [Google Scholar] [CrossRef] [Green Version]
- Saunders, E.C.; Ng, W.W.; Chambers, J.M.; Ng, M.; Naderer, T.; Kromer, J.O.; Likic, V.A.; McConville, M.J. Isotopomer profiling of Leishmania mexicana promastigotes reveals important roles for succinate fermentation and aspartate uptake in tricarboxylic acid cycle (TCA) anaplerosis, glutamate synthesis, and growth. J. Biol. Chem. 2011, 286, 27706–27717. [Google Scholar] [CrossRef] [Green Version]
- McConville, M.J.; de Souza, D.; Saunders, E.; Likic, V.A.; Naderer, T. Living in a phagolysosome; metabolism of Leishmania amastigotes. Trends Parasitol. 2007, 23, 368–375. [Google Scholar] [CrossRef]
- Real, F.; Mortara, R.A. The diverse and dynamic nature of Leishmania parasitophorous vacuoles studied by multidimensional imaging. PLoS Negl. Trop. Dis. 2012, 6, e1518. [Google Scholar] [CrossRef] [Green Version]
- Burchmore, R.J.; Barrett, M.P. Life in vacuoles—Nutrient acquisition by Leishmania amastigotes. Int. J. Parasitol. 2001, 31, 1311–1320. [Google Scholar] [CrossRef]
- Williams, R.A.; Smith, T.K.; Cull, B.; Mottram, J.C.; Coombs, G.H. ATG5 is essential for ATG8-dependent autophagy and mitochondrial homeostasis in Leishmania major. PLoS Pathog. 2012, 8, e1002695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veras, P.S.T.; de Menezes, J.P.B.; Dias, B.R.S. Deciphering the Role Played by Autophagy in Leishmania Infection. Front. Immunol. 2019, 10, 2523. [Google Scholar] [CrossRef] [PubMed]
- Young, J.; Kima, P.E. The Leishmania Parasitophorous Vacuole Membrane at the Parasite-Host Interface. Yale J. Biol. Med. 2019, 92, 511–521. [Google Scholar] [PubMed]
- Yamamoto, Y.H.; Noda, T. Autophagosome formation in relation to the endoplasmic reticulum. J. Biomed. Sci. 2020, 27, 97. [Google Scholar] [CrossRef]
- Ruhland, A.; Leal, N.; Kima, P.E. Leishmania promastigotes activate PI3K/Akt signalling to confer host cell resistance to apoptosis. Cell. Microbiol. 2007, 9, 84–96. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Prasad, S.; Huyghues Despointes, C.E.; Young, J.; Kima, P.E. Leishmania parasitophorous vacuole membranes display phosphoinositides that create conditions for continuous Akt activation and a target for miltefosine in Leishmania infections. Cell. Microbiol. 2018, 20, e12889. [Google Scholar] [CrossRef]
- Ito, K.; Bonora, M.; Ito, K. Metabolism as master of hematopoietic stem cell fate. Int. J. Hematol. 2019, 109, 18–27. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, Y.; Zhang, Y.; Yokota, A.; Yan, X.; Liu, J.; Choi, K.; Li, B.; Sashida, G.; Peng, Y.; Xu, Z.; et al. Pathobiological Pseudohypoxia as a Putative Mechanism Underlying Myelodysplastic Syndromes. Cancer Discov. 2018, 8, 1438–1457. [Google Scholar] [CrossRef] [Green Version]
- Calegari-Silva, T.C.; Vivarini, A.C.; Pereira, R.M.S.; Dias-Teixeira, K.L.; Rath, C.T.; Pacheco, A.S.S.; Silva, G.B.L.; Pinto, C.A.S.; Dos Santos, J.V.; Saliba, A.M.; et al. Leishmania amazonensis downregulates macrophage iNOS expression via Histone Deacetylase 1 (HDAC1): A novel parasite evasion mechanism. Eur. J. Immunol. 2018, 48, 1188–1198. [Google Scholar] [CrossRef] [Green Version]
- Marr, A.K.; MacIsaac, J.L.; Jiang, R.; Airo, A.M.; Kobor, M.S.; McMaster, W.R. Leishmania donovani infection causes distinct epigenetic DNA methylation changes in host macrophages. PLoS Pathog. 2014, 10, e1004419. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Poulaki, A.; Piperaki, E.-T.; Voulgarelis, M. Effects of Visceralising Leishmania on the Spleen, Liver, and Bone Marrow: A Pathophysiological Perspective. Microorganisms 2021, 9, 759. https://doi.org/10.3390/microorganisms9040759
Poulaki A, Piperaki E-T, Voulgarelis M. Effects of Visceralising Leishmania on the Spleen, Liver, and Bone Marrow: A Pathophysiological Perspective. Microorganisms. 2021; 9(4):759. https://doi.org/10.3390/microorganisms9040759
Chicago/Turabian StylePoulaki, Aikaterini, Evangelia-Theophano Piperaki, and Michael Voulgarelis. 2021. "Effects of Visceralising Leishmania on the Spleen, Liver, and Bone Marrow: A Pathophysiological Perspective" Microorganisms 9, no. 4: 759. https://doi.org/10.3390/microorganisms9040759