
SARS-CoV-2 N501Y Introductions and Transmissions in Switzerland from Beginning of October 2020 to February 2021—Implementation of Swiss-Wide Diagnostic Screening and Whole Genome Sequencing

 , , , , , , , , , ,
, , , , , , , , , ,  , , , , , , , and add
Show full author list
, , , , , , , and add
Show full author list
Abstract
:1. Introduction
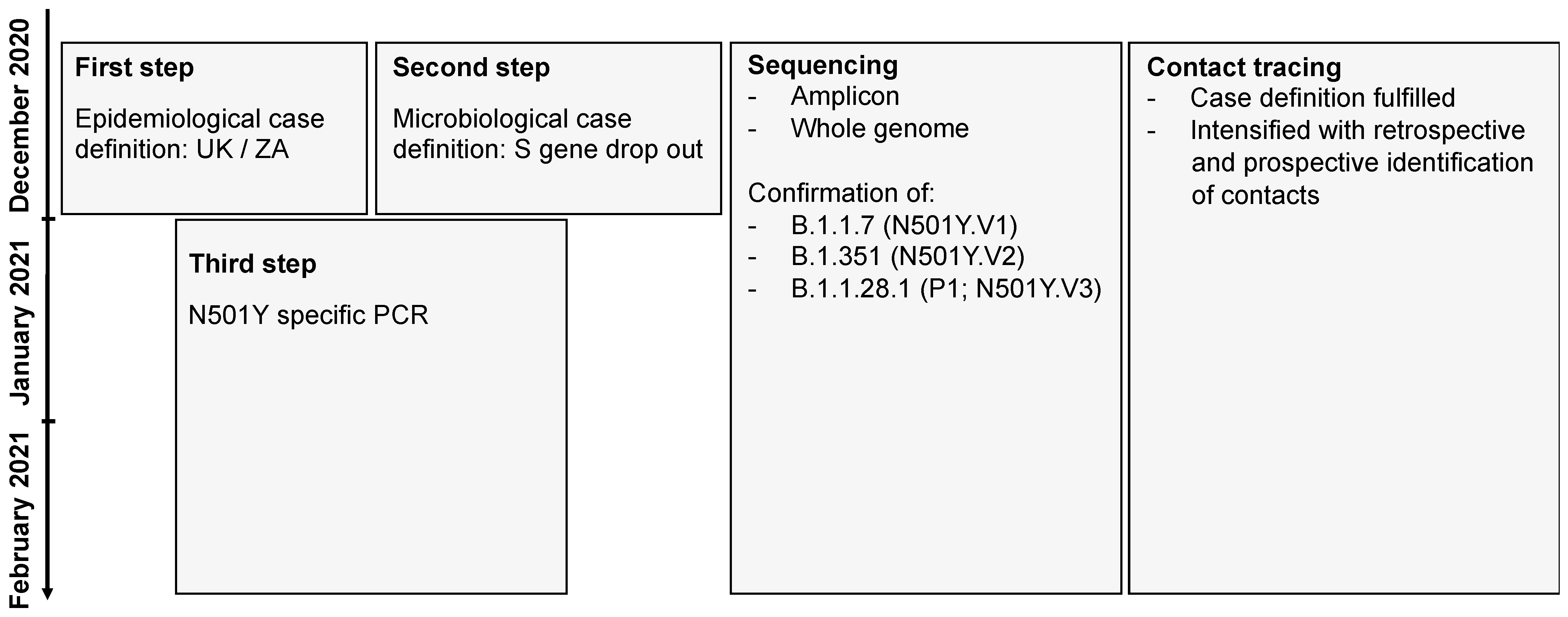
2. Materials and Methods
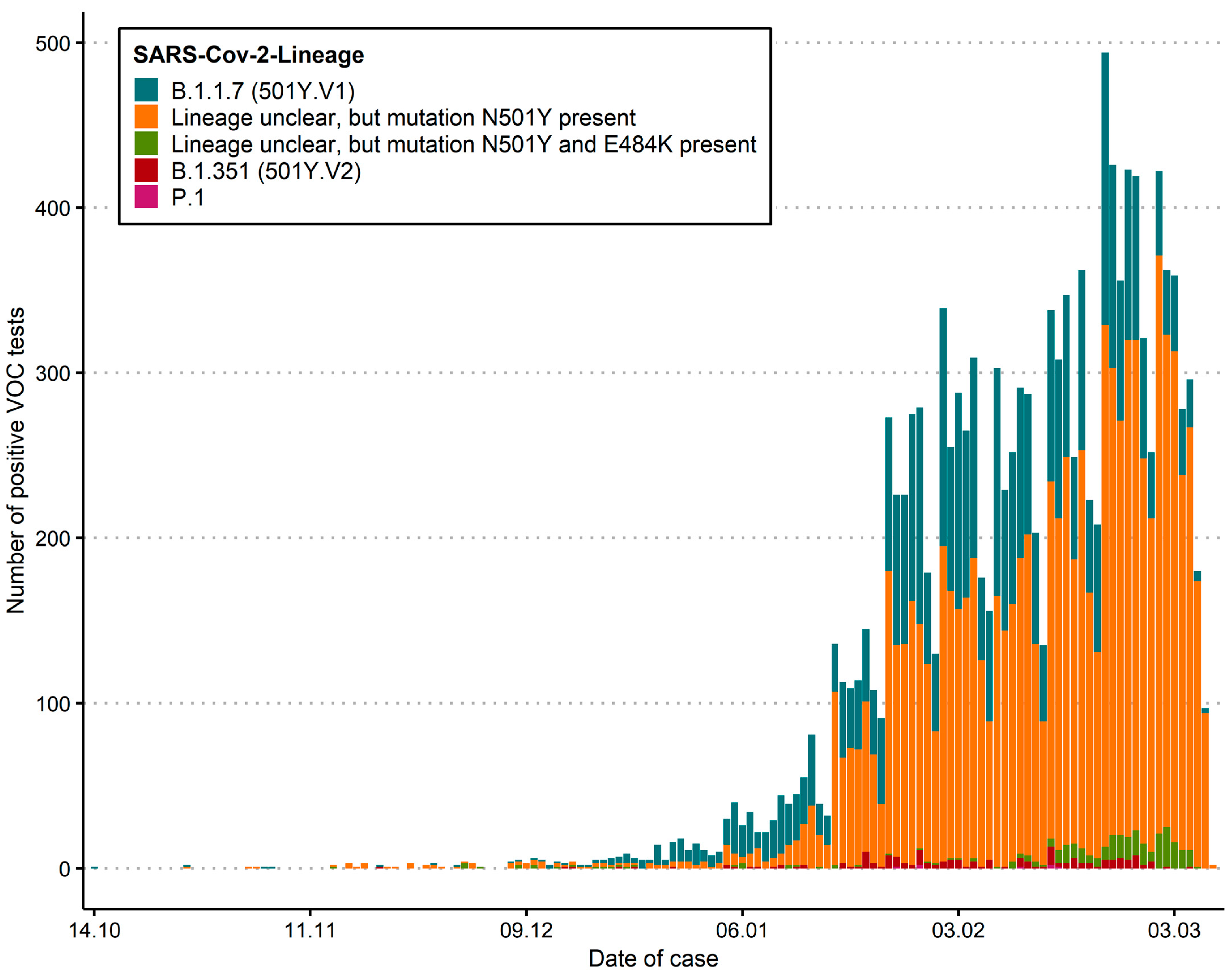
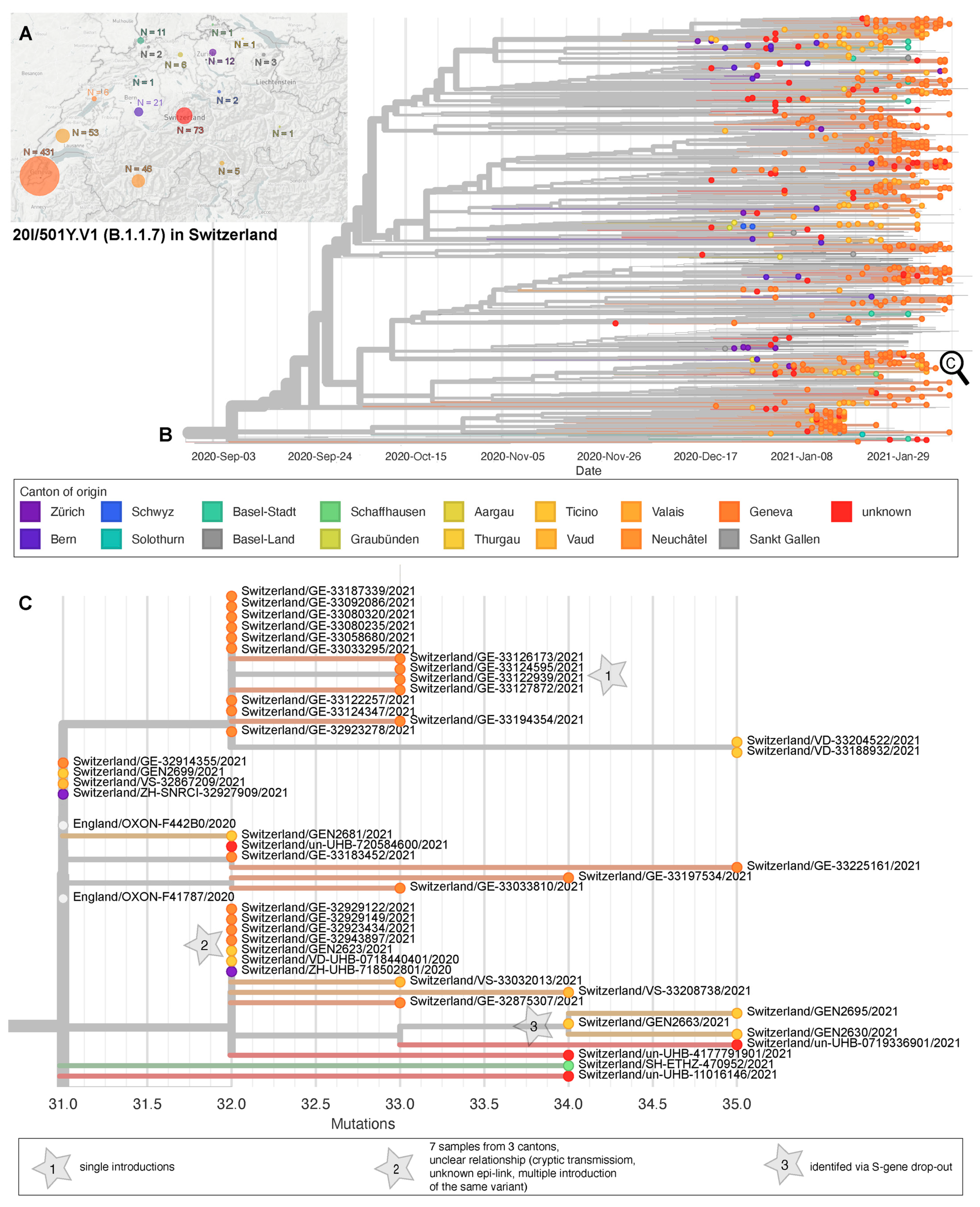
3. Results
3.1. SARS-CoV-2 Case Numbers and Spatio-Temporal Distribution in Switzerland
3.2. Phylogenetic Relatedness of First Cases
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kupferschmidt, K. Fast-spreading U.K. virus variant raises alarms. Science 2021, 371, 9–10. [Google Scholar] [CrossRef] [PubMed]
- Leung, K.; Shum, M.H.; Leung, G.M.; Lam, T.T.; Wu, J.T. Early transmissibility assessment of the N501Y mutant strains of SARS-CoV-2 in the United Kingdom, October to November 2020. Eur. Surveill. 2021, 26, 2002106. [Google Scholar] [CrossRef]
- Tang, J.W.; Tambyah, P.A.; Hui, D.S. Emergence of a new SARS-CoV-2 variant in the UK. J. Infect. 2020. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Li, M.; Ren, R.; Li, L.; Chen, E.Q.; Li, W.; Ying, B. International Expansion of a Novel SARS-CoV-2 Mutant. J. Virol. 2020, 94, e00567-20. [Google Scholar] [CrossRef] [PubMed]
- Davies, N.G.; Abbott, S.; Barnard, R.C.; Jarvis, C.I.; Kucharski, A.J.; Munday, J.; Pearson, C.A.; Russell, T.W.; Tully, D.C.; Washburne, A.D.; et al. Estimated transmissibility and severity of novel SARS-CoV-2 Variant of Concern 202012/01 in England. medRxiv 2020. [Google Scholar] [CrossRef]
- Tegally, H.; Wilkinson, E.; Giovanetti, M.; Iranzadeh, A.; Fonseca, V.; Giandhari, J.; Doolabh, D.; Pillay, S.; San, E.J.; Msomi, N.; et al. Emergence and rapid spread of a new severe acute respiratory syndrome-related coronavirus 2 (SARS-CoV-2) lineage with multiple spike mutations in South Africa. medRxiv 2020. [Google Scholar] [CrossRef]
- Volz, E.; Mishra, S.; Chand, M.; Barrett, J.C.; Johnson, R.; Geidelberg, L.; Hinsley, W.R.; Laydon, D.J.; Dabrera, G.; O’Toole, Á.; et al. Transmission of SARS-CoV-2 Lineage B.1.1.7 in England: Insights from linking epidemiological and genetic data. medRxiv 2021. [Google Scholar] [CrossRef]
- O’Toole, Á.; Hill, V.; Pybus, O.G.; Watts, A.; Bogoch, I.I.; Khan, K.; Messina, J.P. Tracking the International Spread of SARS-CoV-2 Lineages B.1.1.7 and B.1.351/501Y-V2. Virological 2021. Available online: https://virological.org/t/tracking-the-international-spread-of-sars-cov-2-lineages-b-1-1-7-and-b-1-351-501y-v2/592 (accessed on 6 February 2021).
- Voloch, C.M.; da Silva Francisco, R.; de Almeida, L.G.; Cardoso, C.C.; Brustolini, O.J.; Gerber, A.L.; Guimarães, A.P.D.C.; Mariani, D.; da Costa, R.M.; Ferreira, O.C.; et al. Genomic characterization of a novel SARS-CoV-2 lineage from Rio de Janeiro, Brazil. medRxiv 2020. [Google Scholar] [CrossRef]
- Faria, N.R.; Claro, I.M.; Candido, D.; Moyses Franco, L.A.; Andrade, P.S.; Coletti, T.M.; Silva, C.A.; Sales, F.C.; Manuli, E.R.; Aguiar, R.S.; et al. Genomic characterisation of an emergent SARS-CoV-2 lineage in Manaus: Preliminary findings. Virological 2021. Available online: https://virological.org/t/genomic-characterisation-of-an-emergent-sars-cov-2-lineage-in-manaus-preliminary-findings/586 (accessed on 6 February 2021).
- Naveca, F.; Nascimento, V.; Souza, V.; Corado, A.; Nascimento, F.; Silva, G.; Costa, A.; Duarte, D.; Pessoa, K.; Gonçalves, L.; et al. Phylogenetic relationship of SARS-CoV-2 sequences from Amazonas with emerging Brazilian variants harboring mutations E484K and N501Y in the Spike protein. Virological 2021. Available online: https://virological.org/t/phylogenetic-relationship-of-sars-cov-2-sequences-from-amazonas-with-emerging-brazilian-variants-harboring-mutations-e484k-and-n501y-in-the-spike-protein/585 (accessed on 6 February 2021).
- National Institute of Infectious Disease J. Brief Report: New Variant Strain of SARS-CoV-2 Identified in Travelers from Brazil. 2021. Available online: https://www.niid.go.jp/niid/en/2019-ncov-e/10108-covid19-33-en.html (accessed on 6 February 2021).
- Luan, B.; Wang, H.; Huynh, T. Molecular Mechanism of the N501Y Mutation for Enhanced Binding between SARS-CoV-2’s Spike Protein and Human ACE2 Receptor. bioRxiv 2021. [Google Scholar] [CrossRef]
- Kirby, T. New variant of SARS-CoV-2 in UK causes surge of COVID-19. Lancet. Respir. Med. 2021, 9, e20–e21. [Google Scholar] [CrossRef]
- Jahn, K.; Dreifuss, D.; Topolsky, I.; Kull, A.; Ganesanandamoorthy, P.; Fernandez-Cassi, X.; Bänziger, C.; Stachler, E.; Fuhrmann, L.; Jablonski, K.P.; et al. Detection of SARS-CoV-2 variants in Switzerland by genomic analysis of wastewater samples. medRxiv 2021. [Google Scholar] [CrossRef]
- Gupta, R.; Kemp, S.; Harvey, W.; Lytras, S.; Carabelli, A.; Robertson, D. Recurrent independent emergence and transmission of SARS-CoV-2 Spike amino acid H69/V70 deletions. Biol. Sci. 2021. Available online: https://www.researchsquare.com/article/rs-136937/v1 (accessed on 6 February 2021).
- Voloch, C.M.; da Silva Francisco, R.; de Almeida, L.G.; Cardoso, C.C.; Brustolini, O.J.; Gerber, A.L.; Guimarães, A.P.D.C.; Mariani, D.; da Costa, R.M.; Ferreira, O.C.; et al. Preliminary genomic characterisation of an emergent SARS-CoV-2 lineage in the UK defined by a novel set of spike mutations. Virological 2020. Available online: https://virological.org/t/preliminary-genomic-characterisation-of-an-emergent-sars-cov-2-lineage-in-the-uk-defined-by-a-novel-set-of-spike-mutations/563 (accessed on 6 February 2021).
- Isabel, S.; Graña-Miraglia, L.; Gutierrez, J.M.; Bundalovic-Torma, C.; Groves, H.E.; Isabel, M.R.; Eshaghi, A.; Patel, S.N.; Gubbay, J.B.; Poutanen, T.; et al. Evolutionary and structural analyses of SARS-CoV-2 D614G spike protein mutation now documented worldwide. Sci. Rep. 2020, 10, 14031. [Google Scholar] [CrossRef]
- Naqvi, A.A.T.; Fatima, K.; Mohammad, T.; Fatima, U.; Singh, I.K.; Singh, A.; Atif, S.M.; Hariprasad, G.; Hasan, G.M.; Hassan, I. Insights into SARS-CoV-2 genome, structure, evolution, pathogenesis and therapies: Structural genomics approach. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165878. [Google Scholar] [CrossRef]
- Gaebler, C.; Wang, Z.; Lorenzi, J.C.; Muecksch, F.; Finkin, S.; Tokuyama, M.; Cho, A.; Jankovic, M.; Schaefer-Babajew, D.; Oliveira, T.Y.; et al. Evolution of antibody immunity to SARS-CoV-2. Nature 2021, 1–6. [Google Scholar] [CrossRef]
- Guruprasad, L. Human SARS CoV-2 spike protein mutations. Proteins 2021. [Google Scholar] [CrossRef]
- Garry, R.F. Mutations arising in SARS-CoV-2 spike on sustained human-to-human transmission and human-to-animal passage. Virological 2021. Available online: https://virological.org/t/mutations-arising-in-sars-cov-2-spike-on-sustained-human-to-human-transmission-and-human-to-animal-passage/578 (accessed on 6 February 2021).
- Administration UFaD. Genetic Variants of SARS-CoV-2 May Lead to False Negative Results with Molecular Tests for Detection of SARS-CoV-2—Letter to Clinical Laboratory Staff and Health Care Providers. 2021. Available online: https://www.fda.gov/medical-devices/letters-health-care-providers/genetic-variants-sars-cov-2-may-lead-false-negative-results-molecular-tests-detection-sars-cov-2 (accessed on 6 February 2021).
- Control ECfDPa. Risk Assessment: Risk Related to Spread of New SARS-CoV-2 Variants of Concern in the EU/EEA. 2020. Available online: https://www.ecdc.europa.eu/en/publications-data/covid-19-risk-assessment-spread-new-sars-cov-2-variants-eueea (accessed on 6 February 2021).
- Zhang, Y.; Zhang, J.; Chen, Y.; Luo, B.; Yuan, Y.; Huang, F.; Yang, T.; Yu, F.; Liu, J.; Liu, B.; et al. The ORF8 Protein of SARS-CoV-2 Mediates Immune Evasion through Potently Downregulating MHC-I. bioRxiv 2020. [Google Scholar] [CrossRef]
- Weisblum, Y.; Schmidt, F.; Zhang, F.; DaSilva, J.; Poston, D.; Lorenzi, J.C.; Muecksch, F.; Rutkowska, M.; Hoffmann, H.H.; Michailidis, E.; et al. Escape from neutralizing antibodies by SARS-CoV-2 spike protein variants. bioRxiv 2020. [Google Scholar] [CrossRef] [PubMed]
- Greaney, A.J.; Starr, T.N.; Gilchuk, P.; Zost, S.J.; Binshtein, E.; Loes, A.N.; Hilton, S.K.; Huddleston, J.; Eguia, R.; Crawford, K.H.; et al. Complete Mapping of Mutations to the SARS-CoV-2 Spike Receptor-Binding Domain that Escape Antibody Recognition. Cell Host Microbe 2021, 29, 44–57. [Google Scholar] [CrossRef] [PubMed]
- Lauring, A.S.; Hodcroft, E.B. Genetic Variants of SARS-CoV-2-What Do They Mean? JAMA 2021, 325, 529–531. [Google Scholar] [CrossRef]
- Welkers, M.R.A.; Han, A.X.; Reusken, C.; Eggink, D. Possible host-adaptation of SARS-CoV-2 due to improved ACE2 receptor binding in mink. Virus Evol 2021, 7, veaa094. [Google Scholar] [CrossRef]
- Microbiology CCfCMotSSo. Recommendations for Testing Using the N501Y PCR. 2021. Available online: https://www.swissmicrobiology.ch/en/sars-cov-2-pcr-tests (accessed on 6 February 2021).
- Diseases CfEV. Protocol for Specific RT-PCRs for Marker Regions of the Spike Region Indicative of the UK SARS-CoV2 Variant B.1.1.7 and the South African Variant 501Y.V2. 2020. Available online: https://www.hug.ch/sites/interhug/files/structures/laboratoire_de_virologie/protocol_amplification_voc_20201201_uk_geneva.pdf (accessed on 6 February 2021).
- Stange, M.; Mari, A.; Roloff, T.; Seth-Smith, H.M.; Schweitzer, M.; Brunner, M.; Leuzinger, K.; Søgaard, K.K.; Gensch, A.; Tschudin-Sutter, S.; et al. SARS-CoV-2 outbreak in a tri-national urban area is dominated by a B.1 lineage variant linked to mass gathering events. medRxiv 2020. [Google Scholar] [CrossRef]
- Nadeau, S.; Beckmann, C.; Topolsky, I.; Vaughan, T.; Hodcroft, E.; Schaer, T.; Nissen, I.; Santacroce, N.; Burcklen, E.; Ferreira, P.; et al. Quantifying SARS-CoV-2 spread in Switzerland based on genomic sequencing data. medRxiv 2020. [Google Scholar] [CrossRef]
- Gradel, C.; Terrazos Miani, M.A.; Barbani, M.T.; Leib, S.L.; Suter-Riniker, F.; Ramette, A. Rapid and Cost-Efficient Enterovirus Genotyping from Clinical Samples Using Flongle Flow Cells. Genes 2019, 10, 659. [Google Scholar] [CrossRef]
- Neuenschwander, S.M.; Miani, M.A.T.; Amlang, H.; Perroulaz, C.; Bittel, P.; Casanova, C.; Droz, S.; Flandrois, J.P.; Leib, S.L.; Suter-Riniker, F.; et al. A Sample-to-Report Solution for Taxonomic Identification of Cultured Bacteria in the Clinical Setting Based on Nanopore Sequencing. J. Clin. Microbiol 2020, 58. [Google Scholar] [CrossRef]
- Li, H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 2011, 27, 2987–2993. [Google Scholar] [CrossRef] [PubMed]
- Elbe, S.; Buckland-Merrett, G. Data, disease and diplomacy: GISAID’s innovative contribution to global health. Glob. Chall. 2017, 1, 33–46. [Google Scholar] [CrossRef]
- Shu, Y.; McCauley, J. GISAID: Global initiative on sharing all influenza data—From vision to reality. Eur. Surveill. 2017, 22, 30494. [Google Scholar] [CrossRef]
- Hadfield, J.; Megill, C.; Bell, S.M.; Huddleston, J.; Potter, B.; Callender, C.; Sagulenko, P.; Bedford, T. Nextstrain: Real-time tracking of pathogen evolution. Bioinformatics 2018, 34, 4121–4123. [Google Scholar] [CrossRef]
- Leuzinger, K.; Roloff, T.; Gosert, R.; Sogaard, K.; Naegele, K.; Rentsch, K.; Bingisser, R.; Nickel, C.H.; Pargger, H.; Bassetti, S.; et al. Epidemiology of Severe Acute Respiratory Syndrome Coronavirus 2 Emergence Amidst Community-Acquired Respiratory Viruses. J. Infect Dis 2020, 22, 1270–1279. [Google Scholar] [CrossRef]
- Control ECfDPa. Sequencing of SARS-CoV-2—First Update. 2021. Available online: https://www.ecdc.europa.eu/en/publications-data/sequencing-sars-cov-2 (accessed on 6 February 2021).
- Bluhm, A.; Christandl, M.; Gesmundo, F.; Ravn Klausen, F.; Mančinska, L.; Steffan, V.; Stilck França, D.; Werner, A.H. SARS-CoV-2 transmission routes from genetic data: A Danish case study. PLoS ONE 2020, 15, e0241405. [Google Scholar] [CrossRef]
- Correa-Martínez, C.L.; Kampmeier, S.; Kümpers, P.; Schwierzeck, V.; Hennies, M.; Hafezi, W.; Kühn, J.; Pavenstädt, H.; Ludwig, S.; Mellmann, A.; et al. A Pandemic in Times of Global Tourism: Superspreading and Exportation of COVID-19 Cases from a Ski Area in Austria. J. Clin. Microbiol 2020, 58, e0241405. [Google Scholar] [CrossRef] [PubMed]
- Organization WH. Genomic Sequencing of SARS-CoV-2: A Guide to Implementation for Maximum Impact on Public Health. 2021. Available online: https://www.who.int/publications/i/item/9789240018440 (accessed on 6 February 2021).
- Egli, A.; Blanc, D.S.; Greub, G.; Keller, P.M.; Lazarevic, V.; Lebrand, A.; Leib, S.; Neher, R.A.; Perreten, V.; Ramette, A.; et al. Improving the quality and workflow of bacterial genome sequencing and analysis: Paving the way for a Switzerland-wide molecular epidemiological surveillance platform. Swiss Med. Wkly. 2018, 148, w14693. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Canton | B.1.1.7 (501Y.V1) | B.1.351 (501Y.V2) | P.1 | Lineage Not Specified N501Y pos | Lineage Not Specified N501Y and E484K pos | VoC Total |
|---|---|---|---|---|---|---|
| AG | 145 | 3 | 771 | 12 | 931 | |
| AI | 1 | 3 | 4 | |||
| AR | 1 | 54 | 55 | |||
| BE | 528 | 29 | 442 | 33 | 1032 | |
| BL | 259 | 5 | 133 | 1 | 398 | |
| BS | 77 | 3 | 286 | 366 | ||
| FR | 289 | 9 | 1 | 168 | 61 | 528 |
| FL | 32 | 1 | 5 | 1 | 39 | |
| GE | 406 | 11 | 2 | 1432 | 1851 | |
| GL | 6 | 1 | 1 | 10 | 4 | 22 |
| GR | 175 | 2 | 243 | 4 | 424 | |
| JU | 126 | 4 | 28 | 158 | ||
| LU | 24 | 1 | 336 | 42 | 403 | |
| NE | 104 | 331 | 435 | |||
| NW | 3 | 34 | 7 | 44 | ||
| OW | 17 | 17 | ||||
| SG | 146 | 31 | 492 | 6 | 675 | |
| SH | 31 | 10 | 37 | 1 | 79 | |
| SO | 271 | 3 | 141 | 2 | 417 | |
| SZ | 38 | 4 | 124 | 8 | 174 | |
| TG | 68 | 14 | 2 | 329 | 413 | |
| TI | 157 | 2 | 239 | 15 | 413 | |
| UR | 3 | 6 | 2 | 11 | ||
| VD | 726 | 20 | 910 | 9 | 1665 | |
| VS | 199 | 1 | 440 | 640 | ||
| ZG | 8 | 1 | 130 | 10 | 149 | |
| ZH | 372 | 16 | 1 | 1601 | 54 | 2044 |
| CH/FL | 4194 | 172 | 7 | 8742 | 272 | 13387 |
| 25–31 January | 1–7 February | 8–14 February | 15–21 February | 22–28 February | |
|---|---|---|---|---|---|
| Bioanalytica | 6% | 21.2% | 31.1% | 35.9% | 40.5% |
| LMZ Risch | 18.5% | 24.8% | 29% | 48% | 57% |
| University Hospital Basel | 29.5% | 49.3% | 63.3% | 50% | 69.4% |
| University of Bern | 10.2% | 35.9% | 30% | 44.7% | 57.7% |
| University Hospital Geneva | 46.1% | 61.3% | 75.5% | 67.0% | 81.7% |
| University Hospital Lausanne | 30.4% | 51.5% | 53.2% | 65.4% | 81.4% |
| University of Zurich | 20.2% | 34.5% | 36.6% | 46.7% | 65.6% |
| Viollier | 15% | 23.6% | 31.2% | 38% | 61.6% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Goncalves Cabecinhas, A.R.; Roloff, T.; Stange, M.; Bertelli, C.; Huber, M.; Ramette, A.; Chen, C.; Nadeau, S.; Gerth, Y.; Yerly, S.; et al. SARS-CoV-2 N501Y Introductions and Transmissions in Switzerland from Beginning of October 2020 to February 2021—Implementation of Swiss-Wide Diagnostic Screening and Whole Genome Sequencing. Microorganisms 2021, 9, 677. https://doi.org/10.3390/microorganisms9040677
Goncalves Cabecinhas AR, Roloff T, Stange M, Bertelli C, Huber M, Ramette A, Chen C, Nadeau S, Gerth Y, Yerly S, et al. SARS-CoV-2 N501Y Introductions and Transmissions in Switzerland from Beginning of October 2020 to February 2021—Implementation of Swiss-Wide Diagnostic Screening and Whole Genome Sequencing. Microorganisms. 2021; 9(4):677. https://doi.org/10.3390/microorganisms9040677
Chicago/Turabian StyleGoncalves Cabecinhas, Ana Rita, Tim Roloff, Madlen Stange, Claire Bertelli, Michael Huber, Alban Ramette, Chaoran Chen, Sarah Nadeau, Yannick Gerth, Sabine Yerly, and et al. 2021. "SARS-CoV-2 N501Y Introductions and Transmissions in Switzerland from Beginning of October 2020 to February 2021—Implementation of Swiss-Wide Diagnostic Screening and Whole Genome Sequencing" Microorganisms 9, no. 4: 677. https://doi.org/10.3390/microorganisms9040677