
Alteration of Gut Microbiota in Carbapenem-Resistant Enterobacteriaceae Carriers during Fecal Microbiota Transplantation According to Decolonization Periods

 , ,
, ,  , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Subject Selection and FMT Procedures
2.2. Culture Assay and Polymerase Chain Reaction (PCR) for CRE Detection in Carriers
2.3. Quantitative Real-Time PCR and MiSeq Sequencing
2.4. Sequencing Data Analysis
2.5. Co-Occurrence Network Analysis
2.6. Statistical Analysis
3. Results
3.1. Clinical Features of CP-CRE Carriers
3.2. Changes in Gut Microbiota of CP-CRE Carriers after FMT Treatment
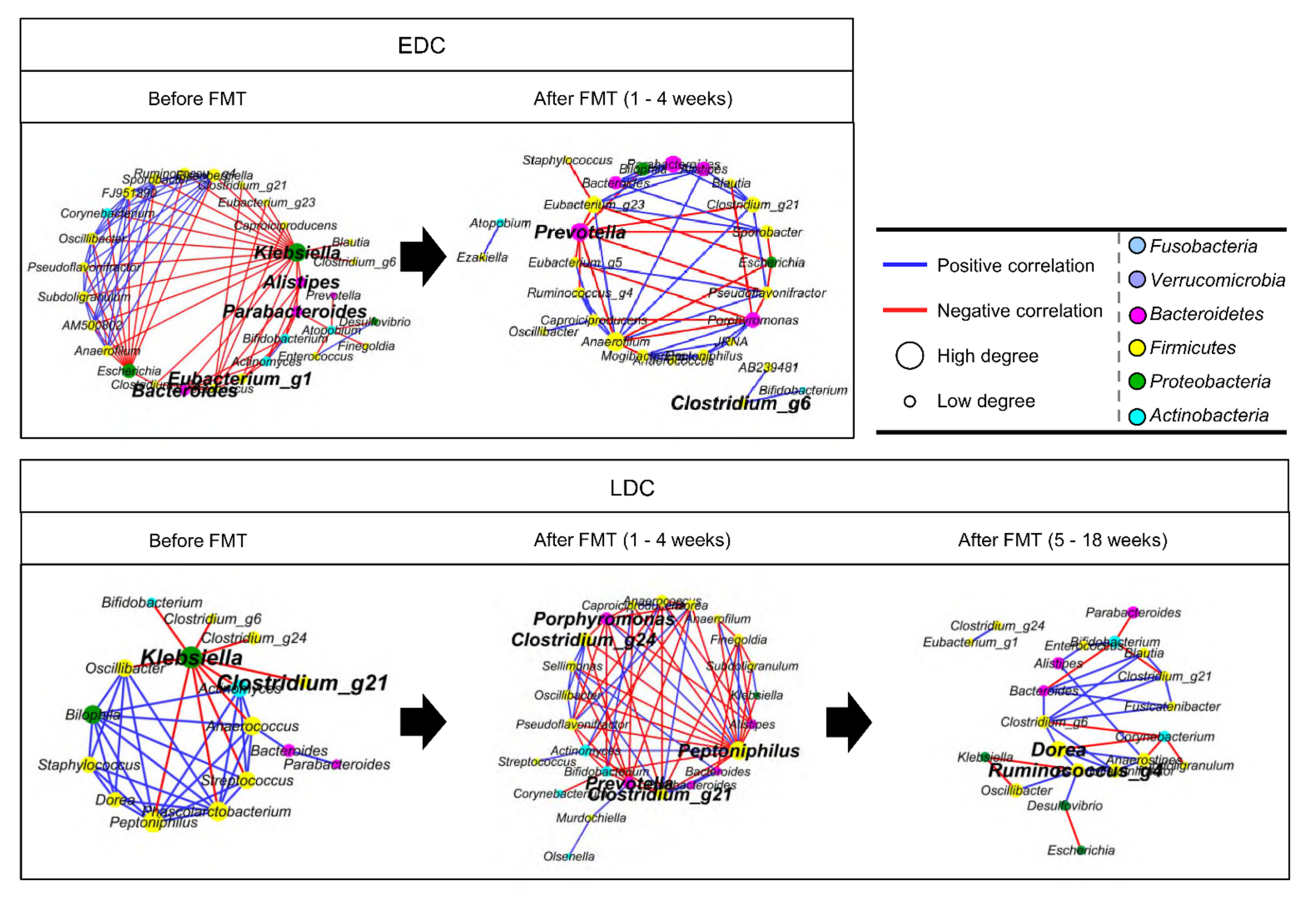
3.3. Comparison of Gut Microbiota between Early Decolonization Carriers and Late Decolonization Carriers
3.4. Different Alteration of Gut Microbiota along Follow-Up Times between EDC and LDC within 4 Weeks after FMT
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bonomo, R.A.; Burd, E.M.; Conly, J.; Limbago, B.M.; Poirel, L.; Segre, J.A.; Westblade, L.F. Carbapenemase-Producing Organisms: A Global Scourge. Clin. Infect. Dis. 2018, 66, 1290–1297. [Google Scholar] [CrossRef] [PubMed]
- Doi, Y.; Paterson, D.L. Carbapenemase-producing Enterobacteriaceae. Semin. Respir. Crit. Care Med. 2015, 36, 74–84. [Google Scholar] [CrossRef] [Green Version]
- Bar-Yoseph, H.; Hussein, K.; Braun, E.; Paul, M. Natural history and decolonization strategies for ESBL/carbapenem-resistant Enterobacteriaceae carriage: Systematic review and meta-analysis. J. Antimicrob. Chemother. 2016, 71, 2729–2739. [Google Scholar] [CrossRef] [PubMed]
- Campos, A.C.; Albiero, J.; Ecker, A.B.; Kuroda, C.M.; Meirelles, L.E.; Polato, A.; Tognim, M.C.; Wingeter, M.A.; Teixeira, J.J. Outbreak of Klebsiella pneumoniae carbapenemase-producing K pneumoniae: A systematic review. Am. J. Infect. Control 2016, 44, 1374–1380. [Google Scholar] [CrossRef]
- Buffie, C.G.; Jarchum, I.; Equinda, M.; Lipuma, L.; Gobourne, A.; Viale, A.; Ubeda, C.; Xavier, J.; Pamer, E.G. Profound alterations of intestinal microbiota following a single dose of clindamycin results in sustained susceptibility to Clostridium difficile-induced colitis. Infect. Immun. 2012, 80, 62–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Debast, S.B.; Bauer, M.P.; Kuijper, E.J.; European Society of Clinical, M.; Infectious, D. European Society of Clinical Microbiology and Infectious Diseases: Update of the treatment guidance document for Clostridium difficile infection. Clin. Microbiol. Infect. 2014, 20 (Suppl. S2), 1–26. [Google Scholar] [CrossRef] [Green Version]
- Millan, B.; Park, H.; Hotte, N.; Mathieu, O.; Burguiere, P.; Tompkins, T.A.; Kao, D.; Madsen, K.L. Fecal Microbial Transplants Reduce Antibiotic-resistant Genes in Patients with Recurrent Clostridium difficile Infection. Clin. Infect. Dis. 2016, 62, 1479–1486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossen, N.G.; Fuentes, S.; van der Spek, M.J.; Tijssen, J.G.; Hartman, J.H.; Duflou, A.; Lowenberg, M.; van den Brink, G.R.; Mathus-Vliegen, E.M.; de Vos, W.M.; et al. Findings From a Randomized Controlled Trial of Fecal Transplantation for Patients with Ulcerative Colitis. Gastroenterology 2015, 149, 110–118.e114. [Google Scholar] [CrossRef]
- Suskind, D.L.; Brittnacher, M.J.; Wahbeh, G.; Shaffer, M.L.; Hayden, H.S.; Qin, X.; Singh, N.; Damman, C.J.; Hager, K.R.; Nielson, H.; et al. Fecal microbial transplant effect on clinical outcomes and fecal microbiome in active Crohn’s disease. Inflamm. Bowel Dis. 2015, 21, 556–563. [Google Scholar] [CrossRef] [Green Version]
- Saha, S.; Tariq, R.; Tosh, P.K.; Pardi, D.S.; Khanna, S. Faecal microbiota transplantation for eradicating carriage of multidrug-resistant organisms: A systematic review. Clin. Microbiol. Infect. 2019, 25, 958–963. [Google Scholar] [CrossRef]
- Manges, A.R.; Steiner, T.S.; Wright, A.J. Fecal microbiota transplantation for the intestinal decolonization of extensively antimicrobial-resistant opportunistic pathogens: A review. Infect. Dis. 2016, 48, 587–592. [Google Scholar] [CrossRef] [PubMed]
- Korach-Rechtman, H.; Hreish, M.; Fried, C.; Gerassy-Vainberg, S.; Azzam, Z.S.; Kashi, Y.; Berger, G. Intestinal Dysbiosis in Carriers of Carbapenem-Resistant Enterobacteriaceae. mSphere 2020, 5, e00173-20. [Google Scholar] [CrossRef]
- Kim, Y.K.; Song, S.A.; Lee, J.N.; Oh, M.; Jo, K.M.; Kim, H.J.; Lee, J.H.; Park, J.; Jang, H.J.; Kim, H.K.; et al. Clinical factors predicting persistent carriage of Klebsiella pneumoniae carbapenemase-producing carbapenem-resistant Enterobacteriaceae among patients with known carriage. J. Hosp. Infect. 2018, 99, 405–412. [Google Scholar] [CrossRef]
- Cammarota, G.; Ianiro, G.; Tilg, H.; Rajilic-Stojanovic, M.; Kump, P.; Satokari, R.; Sokol, H.; Arkkila, P.; Pintus, C.; Hart, A.; et al. European consensus conference on faecal microbiota transplantation in clinical practice. Gut 2017, 66, 569–580. [Google Scholar] [CrossRef] [PubMed]
- Costello, S.P.; Tucker, E.C.; La Brooy, J.; Schoeman, M.N.; Andrews, J.M. Establishing a Fecal Microbiota Transplant Service for the Treatment of Clostridium difficile Infection. Clin. Infect. Dis. 2016, 62, 908–914. [Google Scholar] [CrossRef] [Green Version]
- Terveer, E.M.; van Beurden, Y.H.; Goorhuis, A.; Seegers, J.; Bauer, M.P.; van Nood, E.; Dijkgraaf, M.G.W.; Mulder, C.J.J.; Vandenbroucke-Grauls, C.; Verspaget, H.W.; et al. How to: Establish and run a stool bank. Clin. Microbiol. Infect. 2017, 23, 924–930. [Google Scholar] [CrossRef] [Green Version]
- Division of Human Blood Safety Surveillance. Transfusion Guideline, 3rd ed.; Korean Centers for Disease Control and Prevention: Cheongju, Korea, 2013.
- Clinical & Laboratory Standards Institute. Performance Standards for Antimicrobial Susceptibility Testing. M100S; CLSI: Wayne, PA, USA, 2016. [Google Scholar]
- Morey, K.E.; Vega, R.; Cassidy, P.M.; Buser, G.L.; Rayar, J.K.; Myers, J.A.; Weissman, S.J.; Beldavs, Z.G.; Pfeiffer, C.D. Evaluation of the Carba NP Test in Oregon, 2013. Antimicrob. Agents Chemother. 2017, 61, e3005-15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeong, S.H.; Kim, H.S.; Kim, J.S.; Shin, D.H.; Kim, H.S.; Park, M.J.; Shin, S.; Hong, J.S.; Lee, S.S.; Song, W. Prevalence and Molecular Characteristics of Carbapenemase-Producing Enterobacteriaceae from Five Hospitals in Korea. Ann. Lab. Med. 2016, 36, 529–535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.E.; Lee, J.J.; Lee, M.J.; Kim, B.S. Analysis of microbiome in raw chicken meat from butcher shops and packaged products in South Korea to detect the potential risk of foodborne illness. Food Res. Int. 2019, 122, 517–527. [Google Scholar] [CrossRef] [PubMed]
- Park, J.U.; Oh, B.; Lee, J.P.; Choi, M.H.; Lee, M.J.; Kim, B.S. Influence of Microbiota on Diabetic Foot Wound in Comparison with Adjacent Normal Skin Based on the Clinical Features. BioMed Res. Int. 2019, 2019, 7459236. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoon, S.H.; Ha, S.M.; Kwon, S.; Lim, J.; Kim, Y.; Seo, H.; Chun, J. Introducing EzBioCloud: A taxonomically united database of 16S rRNA gene sequences and whole-genome assemblies. Int. J. Syst. Evol. Microbiol. 2017, 67, 1613–1617. [Google Scholar] [CrossRef]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J.; et al. Introducing mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faust, K.; Raes, J. CoNet app: Inference of biological association networks using Cytoscape. F1000Research 2016, 5, 1519. [Google Scholar] [CrossRef] [PubMed]
- Staley, C.; Kaiser, T.; Vaughn, B.P.; Graiziger, C.T.; Hamilton, M.J.; Rehman, T.U.; Song, K.; Khoruts, A.; Sadowsky, M.J. Predicting recurrence of Clostridium difficile infection following encapsulated fecal microbiota transplantation. Microbiome 2018, 6, 166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ubeda, C.; Taur, Y.; Jenq, R.R.; Equinda, M.J.; Son, T.; Samstein, M.; Viale, A.; Socci, N.D.; van den Brink, M.R.; Kamboj, M.; et al. Vancomycin-resistant Enterococcus domination of intestinal microbiota is enabled by antibiotic treatment in mice and precedes bloodstream invasion in humans. J. Clin. Investig. 2010, 120, 4332–4341. [Google Scholar] [CrossRef]
- Seekatz, A.M.; Aas, J.; Gessert, C.E.; Rubin, T.A.; Saman, D.M.; Bakken, J.S.; Young, V.B. Recovery of the gut microbiome following fecal microbiota transplantation. mBio 2014, 5, e00893-14. [Google Scholar] [CrossRef] [Green Version]
- Weingarden, A.; Gonzalez, A.; Vazquez-Baeza, Y.; Weiss, S.; Humphry, G.; Berg-Lyons, D.; Knights, D.; Unno, T.; Bobr, A.; Kang, J.; et al. Dynamic changes in short- and long-term bacterial composition following fecal microbiota transplantation for recurrent Clostridium difficile infection. Microbiome 2015, 3, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, B.C.; Vatanen, T.; Cutfield, W.S.; O’Sullivan, J.M. The Super-Donor Phenomenon in Fecal Microbiota Transplantation. Front. Cell. Infect. Microbiol. 2019, 9, 2. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Zheng, H.; Lu, R.; Huang, H.; Zhu, H.; Yin, C.; Mo, Y.; Wu, J.; Liu, X.; Deng, M.; et al. Intervening Effects of Total Alkaloids of Corydalis saxicola Bunting on Rats with Antibiotic-Induced Gut Microbiota Dysbiosis Based on 16S rRNA Gene Sequencing and Untargeted Metabolomics Analyses. Front. Microbiol. 2019, 10, 1151. [Google Scholar] [CrossRef]
- Dragomirescu, C.C.; Lixandru, B.E.; Coldea, I.L.; Corneli, O.N.; Pana, M.; Palade, A.M.; Cristea, V.C.; Suciu, I.; Suciu, G.; Manolescu, L.S.C.; et al. Antimicrobial Susceptibility Testing for Corynebacterium Species Isolated from Clinical Samples in Romania. Antibiotics 2020, 9, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pettigrew, M.M.; Gent, J.F.; Kong, Y.; Halpin, A.L.; Pineles, L.; Harris, A.D.; Johnson, J.K. Gastrointestinal Microbiota Disruption and Risk of Colonization with Carbapenem-resistant Pseudomonas aeruginosa in Intensive Care Unit Patients. Clin. Infect. Dis. 2019, 69, 604–613. [Google Scholar] [CrossRef] [PubMed]
- Agudelo-Ochoa, G.M.; Valdes-Duque, B.E.; Giraldo-Giraldo, N.A.; Jaillier-Ramirez, A.M.; Giraldo-Villa, A.; Acevedo-Castano, I.; Yepes-Molina, M.A.; Barbosa-Barbosa, J.; Benitez-Paez, A. Gut microbiota profiles in critically ill patients, potential biomarkers and risk variables for sepsis. Gut Microbes 2020, 12, 1707610. [Google Scholar] [CrossRef] [Green Version]
- Zhu, L.; Liao, R.; Tu, W.; Lu, Y.; Cai, X. Pyrodextrin enhances intestinal function through changing the intestinal microbiota composition and metabolism in early weaned piglets. Appl. Microbiol. Biotechnol. 2020, 104, 4141–4154. [Google Scholar] [CrossRef] [Green Version]
- Engels, C.; Ruscheweyh, H.J.; Beerenwinkel, N.; Lacroix, C.; Schwab, C. The Common Gut Microbe Eubacterium hallii also Contributes to Intestinal Propionate Formation. Front. Microbiol. 2016, 7, 713. [Google Scholar] [CrossRef]
- Trosvik, P.; de Muinck, E.J. Ecology of bacteria in the human gastrointestinal tract--identification of keystone and foundation taxa. Microbiome 2015, 3, 44. [Google Scholar] [CrossRef] [Green Version]
- Comstock, L.E. Importance of glycans to the host-bacteroides mutualism in the mammalian intestine. Cell Host Microbe 2009, 5, 522–526. [Google Scholar] [CrossRef] [Green Version]
- Sequeira, R.P.; McDonald, J.A.K.; Marchesi, J.R.; Clarke, T.B. Commensal Bacteroidetes protect against Klebsiella pneumoniae colonization and transmission through IL-36 signalling. Nat. Microbiol. 2020, 5, 304–313. [Google Scholar] [CrossRef] [PubMed]
- Goloshchapov, O.V.; Olekhnovich, E.I.; Sidorenko, S.V.; Moiseev, I.S.; Kucher, M.A.; Fedorov, D.E.; Pavlenko, A.V.; Manolov, A.I.; Gostev, V.V.; Veselovsky, V.A.; et al. Long-term impact of fecal transplantation in healthy volunteers. BMC Microbiol. 2019, 19, 312. [Google Scholar] [CrossRef] [Green Version]
- Iljazovic, A.; Roy, U.; Galvez, E.J.C.; Lesker, T.R.; Zhao, B.; Gronow, A.; Amend, L.; Will, S.E.; Hofmann, J.D.; Pils, M.C.; et al. Perturbation of the gut microbiome by Prevotella spp. enhances host susceptibility to mucosal inflammation. Mucosal. Immunol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Lopez, J.; Grinspan, A. Fecal Microbiota Transplantation for Inflammatory Bowel Disease. Gastroenterol. Hepatol. 2016, 12, 374–379. [Google Scholar]
- Leszczyszyn, J.J.; Radomski, M.; Leszczyszyn, A.M. Intestinal microbiota transplant—Current state of knowledge. Reumatologia 2016, 54, 24–28. [Google Scholar] [CrossRef] [PubMed]
- Umu, O.C.; Bauerl, C.; Oostindjer, M.; Pope, P.B.; Hernandez, P.E.; Perez-Martinez, G.; Diep, D.B. The Potential of Class II Bacteriocins to Modify Gut Microbiota to Improve Host Health. PLoS ONE 2016, 11, e0164036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andoh, A.; Tsujikawa, T.; Hata, K.; Araki, Y.; Kitoh, K.; Sasaki, M.; Yoshida, T.; Fujiyama, Y. Elevated circulating platelet-derived microparticles in patients with active inflammatory bowel disease. Am. J. Gastroenterol. 2005, 100, 2042–2048. [Google Scholar] [CrossRef]
- Leonetti, D.; Reimund, J.M.; Tesse, A.; Viennot, S.; Martinez, M.C.; Bretagne, A.L.; Andriantsitohaina, R. Circulating microparticles from Crohn’s disease patients cause endothelial and vascular dysfunctions. PLoS ONE 2013, 8, e73088. [Google Scholar] [CrossRef] [Green Version]
- Sweere, J.M.; Van Belleghem, J.D.; Ishak, H.; Bach, M.S.; Popescu, M.; Sunkari, V.; Kaber, G.; Manasherob, R.; Suh, G.A.; Cao, X.; et al. Bacteriophage trigger antiviral immunity and prevent clearance of bacterial infection. Science 2019, 363. [Google Scholar] [CrossRef]
- Rasmussen, T.S.; Mentzel, C.M.J.; Kot, W.; Castro-Mejia, J.L.; Zuffa, S.; Swann, J.R.; Hansen, L.H.; Vogensen, F.K.; Hansen, A.K.; Nielsen, D.S. Faecal virome transplantation decreases symptoms of type 2 diabetes and obesity in a murine model. Gut 2020, 69, 2122–2130. [Google Scholar] [CrossRef]
- Duan, Y.; Llorente, C.; Lang, S.; Brandl, K.; Chu, H.; Jiang, L.; White, R.C.; Clarke, T.H.; Nguyen, K.; Torralba, M.; et al. Bacteriophage targeting of gut bacterium attenuates alcoholic liver disease. Nature 2019, 575, 505–511. [Google Scholar] [CrossRef]
- Wong, W.F.; Santiago, M. Microbial approaches for targeting antibiotic-resistant bacteria. Microb. Biotechnol. 2017, 10, 1047–1053. [Google Scholar] [CrossRef] [Green Version]
- Jouhten, H.; Mattila, E.; Arkkila, P.; Satokari, R. Reduction of Antibiotic Resistance Genes in Intestinal Microbiota of Patients with Recurrent Clostridium difficile Infection after Fecal Microbiota Transplantation. Clin. Infect. Dis. 2016, 63, 710–711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bilinski, J.; Grzesiowski, P.; Sorensen, N.; Madry, K.; Muszynski, J.; Robak, K.; Wroblewska, M.; Dzieciatkowski, T.; Dulny, G.; Dwilewicz-Trojaczek, J.; et al. Fecal Microbiota Transplantation in Patients with Blood Disorders Inhibits Gut Colonization with Antibiotic-Resistant Bacteria: Results of a Prospective, Single-Center Study. Clin. Infect. Dis. 2017, 65, 364–370. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Carrier No. | Age | Sex | CRE Type | Duration of CRE Carriage before FMT | Carbapenem Use ≥ 3 days after CRE Carriage | CRE (+) in Clinical Specimen after CRE Carriage | Concurrent C. difficile Infection after CRE Carriage | Prolonged Hospitalization ≥2 Months after CRE Carriage | FMT Material | FMT Procedure | Time to Decolonization of CRE from FMT |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 78 | Female | KPC-CRE | 7 months | + | + | - | + | Unrelated donor, frozen stool | Terminal ileum and ascending colon via colonoscopy | 25 days |
| 2 | 68 | Male | KPC-CRE | 4 months | + | + | - | + | Unrelated donor, frozen stool | Duodenum via EGD and ascending colon via colonoscopy | 26 days |
| 3 | 80 | Female | KPC-CRE | 7 months | + | + | - | + | Unrelated donor, frozen stool | Duodenum via EGD (1st and 2nd FMT) | 15 days after 2nd FMT (106 days after 1st FMT) |
| 4 | 79 | Male | KPC-CRE | 6 months | + | + | - | + | Unrelated donor, frozen stool | Terminal ileum via colonoscopy | 51 days |
| 5 | 75 | Female | KPC-CRE (& VRE) | 5 months | + | + | + | + | Unrelated donor, frozen stool | Ascending colon via colonoscopy | 15 days |
| 6 | 75 | Female | KPC-CRE (& VRE) | 4 months | + | + | - | + | Unrelated donor, frozen stool | Terminal ileum via colonoscopy (1st FMT), duodenum via EGD (2nd FMT) | 34 days after 2nd FMT (117 days after 1st FMT) |
| 7 | 57 | Male | KPC-CRE | 7 months | + | - | - | + | Unrelated donor, frozen stool | Terminal ileum and ascending colon via colonoscopy (1st and 2nd FMT) | not decolonized (followed until 138 days after 1st FMT) |
| 8 | 81 | Female | KPC-CRE | 10 months | + | + | + | + | Unrelated donor, frozen and capsulized stool | Duodenum via EGD (1st and 2nd FMT), 20 capsules daily for 2 days (3rd FMT) | 16 days after 3rd FMT (137 days after 1st FMT) |
| 9 | 65 | Female | KPC-CRE | 3 months | + | - | - | + | Unrelated donor, frozen stool | Terminal ileum via colonoscopy (1st FMT) duodenum via EGD (2nd FMT) | 16 days after 2nd FMT (92 days after 1st FMT) |
| 10 | 69 | Female | KPC-CRE | 4 months | + | + | - | + | Unrelated donor, frozen stool | Terminal ileum and ascending colon via colonoscopy | 18 days |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, J.-J.; Yong, D.; Suk, K.T.; Kim, D.J.; Woo, H.-J.; Lee, S.S.; Kim, B.-S. Alteration of Gut Microbiota in Carbapenem-Resistant Enterobacteriaceae Carriers during Fecal Microbiota Transplantation According to Decolonization Periods. Microorganisms 2021, 9, 352. https://doi.org/10.3390/microorganisms9020352
Lee J-J, Yong D, Suk KT, Kim DJ, Woo H-J, Lee SS, Kim B-S. Alteration of Gut Microbiota in Carbapenem-Resistant Enterobacteriaceae Carriers during Fecal Microbiota Transplantation According to Decolonization Periods. Microorganisms. 2021; 9(2):352. https://doi.org/10.3390/microorganisms9020352
Chicago/Turabian StyleLee, Jin-Jae, Dongeun Yong, Ki Tae Suk, Dong Joon Kim, Heung-Jeong Woo, Seung Soon Lee, and Bong-Soo Kim. 2021. "Alteration of Gut Microbiota in Carbapenem-Resistant Enterobacteriaceae Carriers during Fecal Microbiota Transplantation According to Decolonization Periods" Microorganisms 9, no. 2: 352. https://doi.org/10.3390/microorganisms9020352