
The Oral Bacterial Community in Melanophryniscus admirabilis (Admirable Red-Belly Toads): Implications for Conservation

 ,
,  , ,
, ,  ,
, 
Abstract
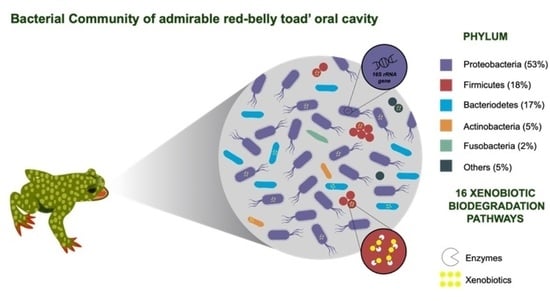
:
1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. Ethics and Sampling Permits
2.3. DNA Extraction, PCR-Amplification of Bacterial 16S rRNA Genes and Sequencing
2.4. Bacterial Community Analysis
2.5. Functional Predictions from Amplicon Sequences
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Frost, D.R. Amphibian Species of the World: An Online Reference. Version 6.1. Available online: https://amphibiansoftheworld.amnh.org/index.php (accessed on 8 January 2021).
- Preuss, J.F.; Greenspan, S.E.; Rossi, E.M.; Gonsales, E.M.L.; Neely, W.J.; Valiati, V.H.; Woodhams, D.C.; Becker, C.G.; Tozetti, A.M. Widespread pig farming practice linked to shifts in skin microbiomes and disease in pond-breeding amphibians. Environ. Sci. Technol. 2020, 54, 11301–11312. [Google Scholar] [CrossRef]
- IUCN 2020. The IUCN Red List of Threatened Species. Version 2020-3. Available online: https://www.iucnredlist.org (accessed on 8 January 2021).
- Zank, C.; Becker, F.G.; Abadie, M.; Baldo, D.; Maneyro, R.; Borges-Martins, M. Climate change and the distribution of neotropical red-bellied toads (Melanophryniscus, Anura, Amphibia): How to prioritize species and populations? PLoS ONE 2014, 9, e94625. [Google Scholar] [CrossRef] [PubMed]
- Bordignon, D.W.; Caorsi, V.Z.; Colombo, P.; Abadie, M.; Brack, I.V.; Dasoler, B.T.; Borges-Martins, M. Are the unken reflex and the aposematic colouration of Red-Bellied Toads efficient against bird predation? PLoS ONE 2018, 13, e0193551. [Google Scholar] [CrossRef] [Green Version]
- Di-Bernardo, M.; Maneyro, R.; Grillo, H. New species of Melanophryniscus (Anura: Bufonidae) from Rio Grande do Sul, Southern Brazil. South Am. J. Herpetol. 2006, 40, 261–266. [Google Scholar] [CrossRef]
- Daly, J.W.; Highet, R.J.; Myers, C.W. Occurrence of skin alkaloids in non-dendrobatid frogs from Brazil (Bufonidae), Australia (Myobatrachidae) and Madagascar (Mantellinae). Toxicon 1984, 22, 905–919. [Google Scholar] [CrossRef]
- Garraffo, H.M.; Andriamaharavo, N.R.; Vaira, M.; Quiroga, M.F.; Heit, C.; Spande, T.F. Alkaloids from single skins of the Argentinian toad Melanophryniscus rubriventris (ANURA, BUFONIDAE): An unexpected variability in alkaloid profiles and a profusion of new structures. SpringerPlus 2012, 1, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daly, J.W.; Spande, T.F.; Garraffo, H.M. Alkaloids from amphibian skin: A tabulation of over eight-hundred compounds. J. Nat. Prod. 2005, 68, 1556–1575. [Google Scholar] [CrossRef]
- Hantak, M.M.; Grant, T.; Reinsch, S.; McGinnity, D.; Loring, M.; Toyooka, N.; Saporito, R.A. Dietary alkaloid sequestration in a poison frog: An experimental test of alkaloid uptake in Melanophryniscus stelzneri (Bufonidae). J. Chem. Ecol. 2013, 39, 1400–1406. [Google Scholar] [CrossRef]
- Saporito, R.A.; Donnelly, M.A.; Spande, T.F.; Garraffo, H.M. A review of chemical ecology in poison frogs. Chemoecology 2012, 22, 159–168. [Google Scholar] [CrossRef]
- Fonte, L.; Abadie, M.; Mendes, T.; Zank, C.; Borges-Martins, M. The times they are a-changing: How a multi-institutional effort stopped the construction of a hydroelectric power plant that threatened a critically endangered red-belly toad in Southern Brazil. FrogLog 2014, 22, 18–21. [Google Scholar]
- Da Silva, P.R.; Borges-Martins, M.; Oliveira, G.T. Melanophryniscus admirabilis tadpoles’ responses to sulfentrazone and glyphosate-based herbicides: An approach on metabolism and antioxidant defenses. Environ. Sci. Pollut. Res. 2020, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Karl, J.P.; Hatch, A.M.; Arcidiacono, S.M.; Pearce, S.C.; Pantoja-Feliciano, I.G.; Doherty, L.A.; Soares, J.W. effects of psychological, environmental and physical stressors on the gut microbiota. Front. Microbiol. 2018, 9, 2013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stevenson, T.J.; Duddleston, K.N.; Buck, C.L. Effects of season and host physiological state on the diversity, density, and activity of the arctic ground squirrel cecal microbiota. Appl. Environ. Microbiol. 2014, 80, 5611–5622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolaños, L.M.; Rosenblueth, M.; Castillo-Ramírez, S.; Figuier-Huttin, G.; Martínez-Romero, E. Species-specific diversity of novel bacterial lineages and differential abundance of predicted pathways for toxic compound degradation in scorpion gut microbiota. Environ. Microbiol. 2016, 18, 1364–1378. [Google Scholar] [CrossRef]
- Shu, Y.; Hong, P.; Tang, D.; Qing, H.; Donde, O.O.; Wang, H.; Xiao, B.; Shu, Y. Comparison of intestinal microbes in female and male Chinese concave-eared frogs (Odorrana tormota) and effect of nematode infection on gut bacterial communities. Microbiologyopen 2019, 8, e00749. [Google Scholar] [CrossRef] [Green Version]
- Kueneman, J.G.; Weiss, S.; McKenzie, V.J. Composition of micro-eukaryotes on the skin of the cascades frog (Rana cascadae) and patterns of correlation between skin microbes and Batrachochytrium dendrobatidis. Front. Microbiol. 2017, 8, 2350. [Google Scholar] [CrossRef]
- Jiménez, R.R.; Sommer, S. The amphibian microbiome: Natural range of variation, pathogenic dysbiosis, and role in conservation. Biodivers. Conserv. 2017, 26, 763–786. [Google Scholar] [CrossRef]
- Machado, A.B.M.; Drummond, G.M.; Paglia, A.P. Livro Vermelho da Fauna Brasileira Ameaçada de Extinção; MMA: Brasília, Brazil, 2008. [Google Scholar]
- Schulte, U.; Gebhard, F.; Heinz, L.; Veith, M.; Hochkirch, A. Buccal swabs as a reliable non-invasive tissue sampling method for DNA analysis in the lacertid lizard Podarcis muralis. North. West. J. Zool. 2011, 7, 325–328. [Google Scholar]
- Caporaso, J.G.; Lauber, C.L.; Walters, W.A.; Berg-Lyons, D.; Huntley, J.; Fierer, N.; Owens, S.M.; Betley, J.; Fraser, L.; Bauer, M.; et al. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J. 2012, 6, 1621–1624. [Google Scholar] [CrossRef] [Green Version]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [Green Version]
- Katoh, K.; Misawa, K.I.; Kuma, K.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bokulich, N.A.; Kaehler, B.D.; Rideout, J.R.; Dillon, M.; Bolyen, E.; Knight, R.; Huttley, G.; Caporaso, J.G. Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2’s q2-feature-classifier plugin. Microbiome 2018, 6, 1–17. [Google Scholar] [CrossRef] [PubMed]
- McMurdie, P.J.; Holmes, S. phyloseq: An R Package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lahti, L.; Shetty, S.; Blake, T.; Salojarvi, J. Microbiome: Tools for Microbiome Analysis in r. 2017. Available online: http://microbiome.github.com/microbiome (accessed on 8 January 2021).
- Wilcoxon, F. Individual Comparisons by Ranking Methods. Biom. Bull. 1945, 1, 80. [Google Scholar] [CrossRef]
- Douglas, G.M.; Maffei, V.J.; Zaneveld, J.; Yurgel, S.N.; Brown, J.R.; Taylor, C.M.; Huttenhower, C.; Langille, M.G.I. PICRUSt2: An improved and customizable approach for metagenome inference. BioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Kanehisa, M.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 2016, 44, D457–D462. [Google Scholar] [CrossRef] [Green Version]
- Parks, D.H.; Tyson, G.W.; Hugenholtz, P.; Beiko, R.G. STAMP: Statistical analysis of taxonomic and functional profiles. Bioinformatic 2014, 30, 3123–3124. [Google Scholar] [CrossRef] [Green Version]
- Kueneman, J.G.; Bletz, M.C.; McKenzie, V.J.; Becker, C.G.; Joseph, M.B.; Abarca, J.G.; Archer, H.; Arellano, A.L.; Bataille, A.; Becker, M.; et al. Community richness of amphibian skin bacteria correlates with bioclimate at the global scale. Nat. Ecol. Evol. 2019, 3, 381–389. [Google Scholar] [CrossRef]
- Chang, C.-W.; Huang, B.-H.; Lin, S.-M.; Huang, C.-L.; Liao, P.-C. Changes of diet and dominant intestinal microbes in farmland frogs. BMC Microbiol. 2016, 16, 33. [Google Scholar] [CrossRef] [Green Version]
- Scheele, B.C.; Pasmans, F.; Skerratt, L.F.; Berger, L.; Martel, A.; Beukema, W.; Acevedo, A.A.; Burrowes, P.A.; Carvalho, T.; Catenazzi, A.; et al. Amphibian fungal panzootic causes catastrophic and ongoing loss of biodiversity. Science 2019, 363, 1459–1463. [Google Scholar] [CrossRef]
- Bletz, M.C.; Goedbloed, D.J.; Sanchez, E.; Reinhardt, T.; Tebbe, C.C.; Bhuju, S.; Geffers, R.; Jarek, M.; Vences, M.; Steinfartz, S. Amphibian gut microbiota shifts differentially in community structure but converges on habitat-specific predicted functions. Nat. Commun. 2016, 7, 13699. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.-H.; Chang, C.-W.; Huang, C.-W.; Gao, J.; Liao, P.-C. Composition and functional specialists of the gut microbiota of frogs reflect habitat differences and agricultural activity. Front. Microbiol. 2018, 8, 2670. [Google Scholar] [CrossRef] [PubMed]
- Kohl, K.D.; Cary, T.L.; Karasov, W.H.; Dearing, M.D. Restructuring of the amphibian gut microbiota through metamorphosis. Environ. Microbiol. Rep. 2013, 5, 899–903. [Google Scholar] [CrossRef] [PubMed]
- Yun, J.-H.; Roh, S.W.; Whon, T.W.; Jung, M.-J.; Kim, M.-S.; Park, D.-S.; Yoon, C.; Nam, Y.-D.; Kim, Y.-J.; Choi, J.-H.; et al. Insect gut bacterial diversity determined by environmental habitat, diet, developmental stage, and phylogeny of host. Appl. Environ. Microbiol. 2014, 80, 5254–5264. [Google Scholar] [CrossRef] [Green Version]
- Itävaara, M.; Salavirta, H.; Marjamaa, K.; Ruskeeniemi, T. Geomicrobiology and metagenomics of terrestrial deep subsurface microbiomes. Adv. Clin. Chem. 2016, 94, 1–77. [Google Scholar] [CrossRef]
- Ramsey, J.P.; Mercurio, A.; Holland, J.A.; Harris, R.N.; Minbiole, K. The cutaneous bacterium Janthinobacterium lividum inhibits the growth of Trichophyton rubrum in vitro. Int. J. Dermatol. 2015, 54, 156–159. [Google Scholar] [CrossRef]
- Lam, B.A.; Walke, J.B.; Vredenburg, V.T.; Harris, R.N. Proportion of individuals with anti-Batrachochytrium dendrobatidis skin bacteria is associated with population persistence in the frog Rana muscosa. Biol. Conserv. 2010, 143, 529–531. [Google Scholar] [CrossRef]
- Banas, J.A.; Loesche, W.J.; Nace, G.W. Classification and distribution of large intestinal bacteria in nonhibernating and hibernating leopard frogs (Rana pipiens). Appl. Environ. Microbiol. 1988, 54, 2305–2310. [Google Scholar] [CrossRef] [Green Version]
- Christian, K.; Weitzman, C.; Rose, A.; Kaestli, M.; Gibb, K. Ecological patterns in the skin microbiota of frogs from tropical Australia. Ecol. Evol. 2018, 8, 10510–10519. [Google Scholar] [CrossRef] [Green Version]
- Depoorter, E.; Bull, M.J.; Peeters, C.; Coenye, T.; Vandamme, P.; Mahenthiralingam, E. Burkholderia: An update on taxonomy and biotechnological potential as antibiotic producers. Appl. Microbiol. Biotechnol. 2016, 100, 5215–5229. [Google Scholar] [CrossRef]
- Willems, A.; De Ley, J.; Gillis, M.; Kersters, K. NOTES: Comamonadaceae, a new family encompassing the acidovorans rRNA complex, including Variovorax paradoxus gen. nov., comb. nov., for Alcaligenes paradoxus (Davis 1969). Int. J. Syst. Bacteriol. 1991, 41, 445–450. [Google Scholar] [CrossRef] [Green Version]
- Noel, A.C.; Guo, H.-Y.; Mandica, M.; Hu, D.L. Frogs use a viscoelastic tongue and non-Newtonian saliva to catch prey. J. R. Soc. Interface 2017, 14, 20160764. [Google Scholar] [CrossRef] [PubMed]
- Lima, T.M.F. Descrição da Dieta de Melanophryniscus Admirabilis (Anura: Bufonidae). Bachelor’s Thesis, Universidade Federal do Rio Grande do Sul, Porto Alegre, Brasil, 2014. [Google Scholar]
- Trevelline, B.K.; Fontaine, S.S.; Hartup, B.K.; Kohl, K.D. Conservation biology needs a microbial renaissance: A call for the consideration of host-associated microbiota in wildlife management practices. Proc. R. Soc. B Boil. Sci. 2019, 286, 20182448. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SAMPLE (ID) | SVL * (mm) | MASS (g) | SEX |
|---|---|---|---|
| AC421 | 31.51 | 3.1 | Male |
| AC422 | 29.88 | 3 | Male |
| AC423 | 30.63 | 3.2 | Male |
| TA01 I | 35.46 | 3.6 | Female |
| TA02 II | 33.62 | 3.2 | Male |
| TA04 IV | 31.50 | 2.7 | Male |
| TA05 | 34.72 | 3.8 | Female |
| TA07 VII | 32.33 | 3.6 | Male |
| TA10 X | 33.87 | 4.2 | Female |
| TA11 XI | 33.67 | 4.1 | Male |
| TA12 XII | 35.07 | 3.9 | Male |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mann, M.B.; Prichula, J.; de Castro, Í.M.S.; Severo, J.M.; Abadie, M.; De Freitas Lima, T.M.; Caorsi, V.; Borges-Martins, M.; Frazzon, J.; Frazzon, A.P.G. The Oral Bacterial Community in Melanophryniscus admirabilis (Admirable Red-Belly Toads): Implications for Conservation. Microorganisms 2021, 9, 220. https://doi.org/10.3390/microorganisms9020220
Mann MB, Prichula J, de Castro ÍMS, Severo JM, Abadie M, De Freitas Lima TM, Caorsi V, Borges-Martins M, Frazzon J, Frazzon APG. The Oral Bacterial Community in Melanophryniscus admirabilis (Admirable Red-Belly Toads): Implications for Conservation. Microorganisms. 2021; 9(2):220. https://doi.org/10.3390/microorganisms9020220
Chicago/Turabian StyleMann, Michele Bertoni, Janira Prichula, Ícaro Maia Santos de Castro, Juliana Mello Severo, Michelle Abadie, Thayná Mendes De Freitas Lima, Valentina Caorsi, Márcio Borges-Martins, Jeverson Frazzon, and Ana Paula Guedes Frazzon. 2021. "The Oral Bacterial Community in Melanophryniscus admirabilis (Admirable Red-Belly Toads): Implications for Conservation" Microorganisms 9, no. 2: 220. https://doi.org/10.3390/microorganisms9020220