
Diverse Single-Stranded DNA Viruses Identified in Chicken Buccal Swabs

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection and Processing
2.2. Illumina MiSeq Sequencing
2.3. Identification of DNA Viruses and Determination of Complete Viral Genomes
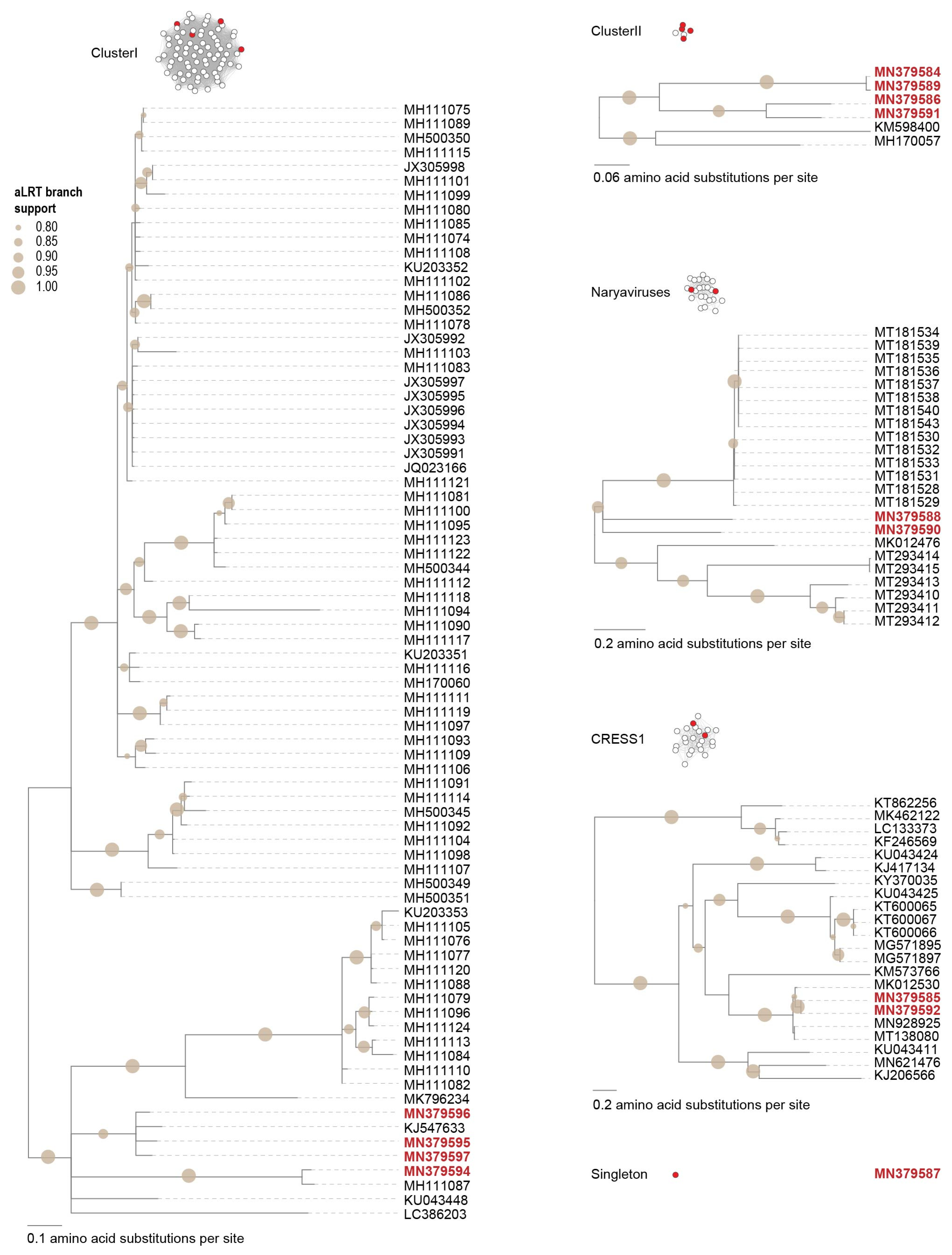
2.4. Sequence Similarity Network Analysis
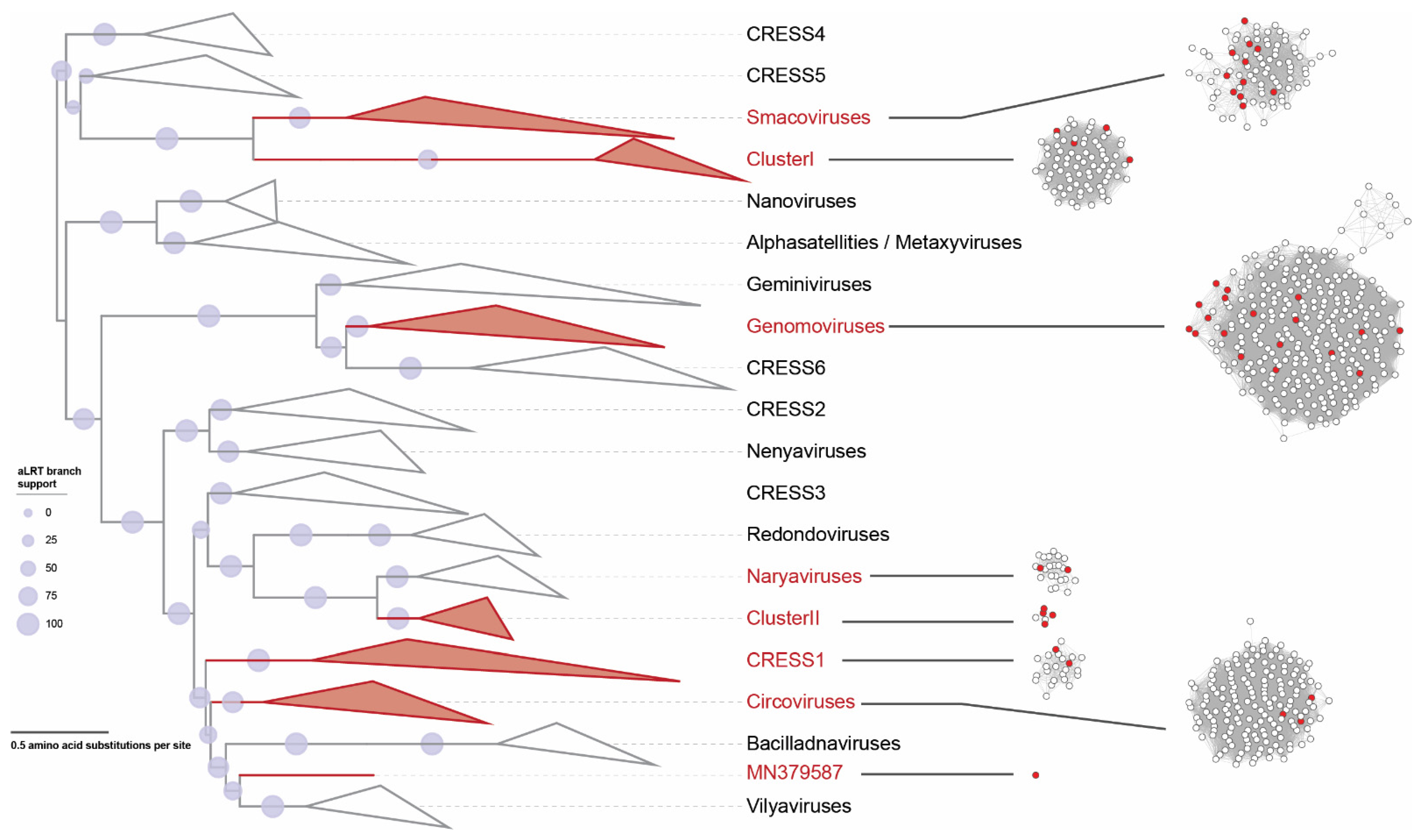
2.5. Phylogenetic Analyses of the Cressdnaviruses
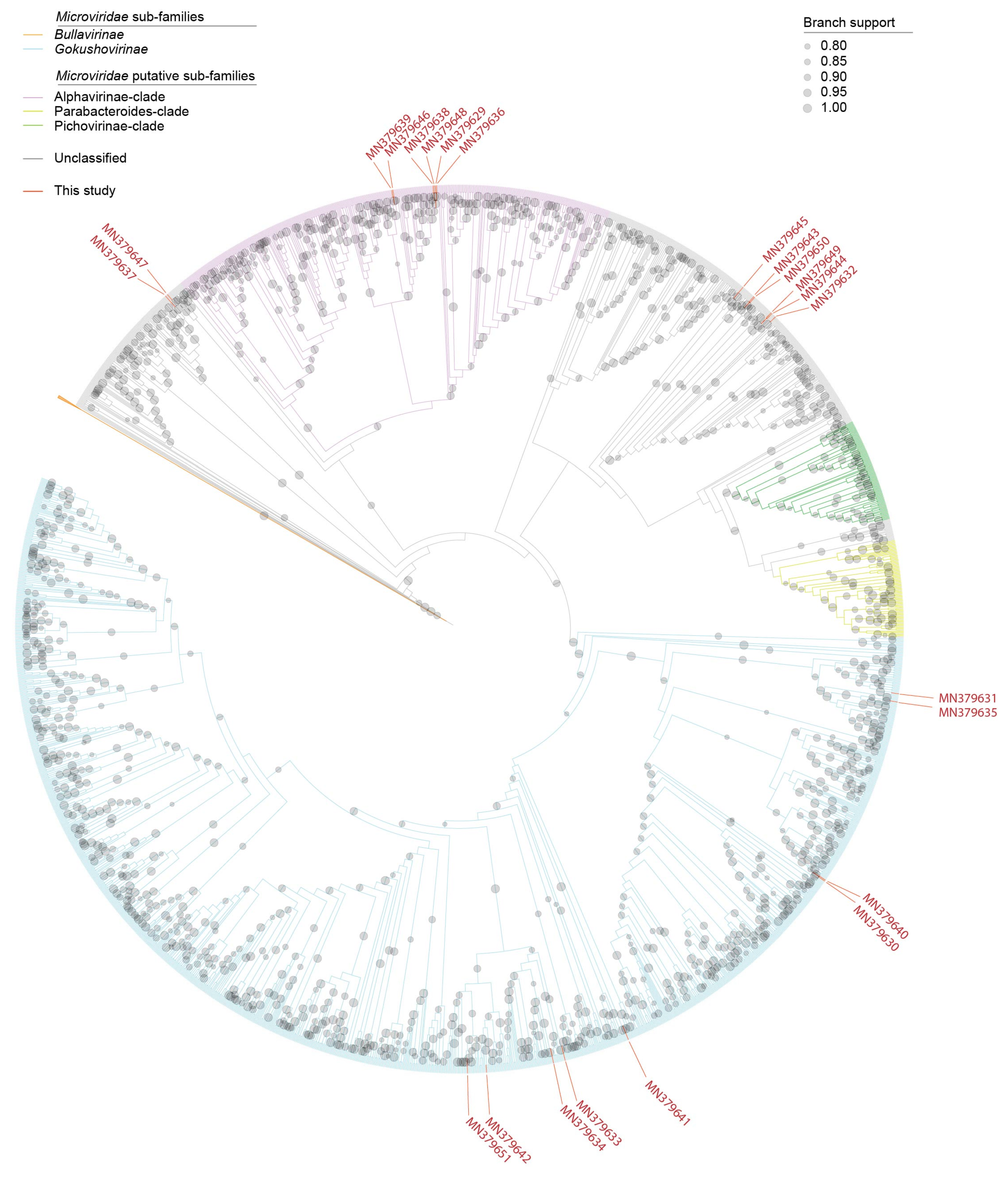
2.6. Phylogenetic Analyses of the Microviruses
3. Results and Discussion
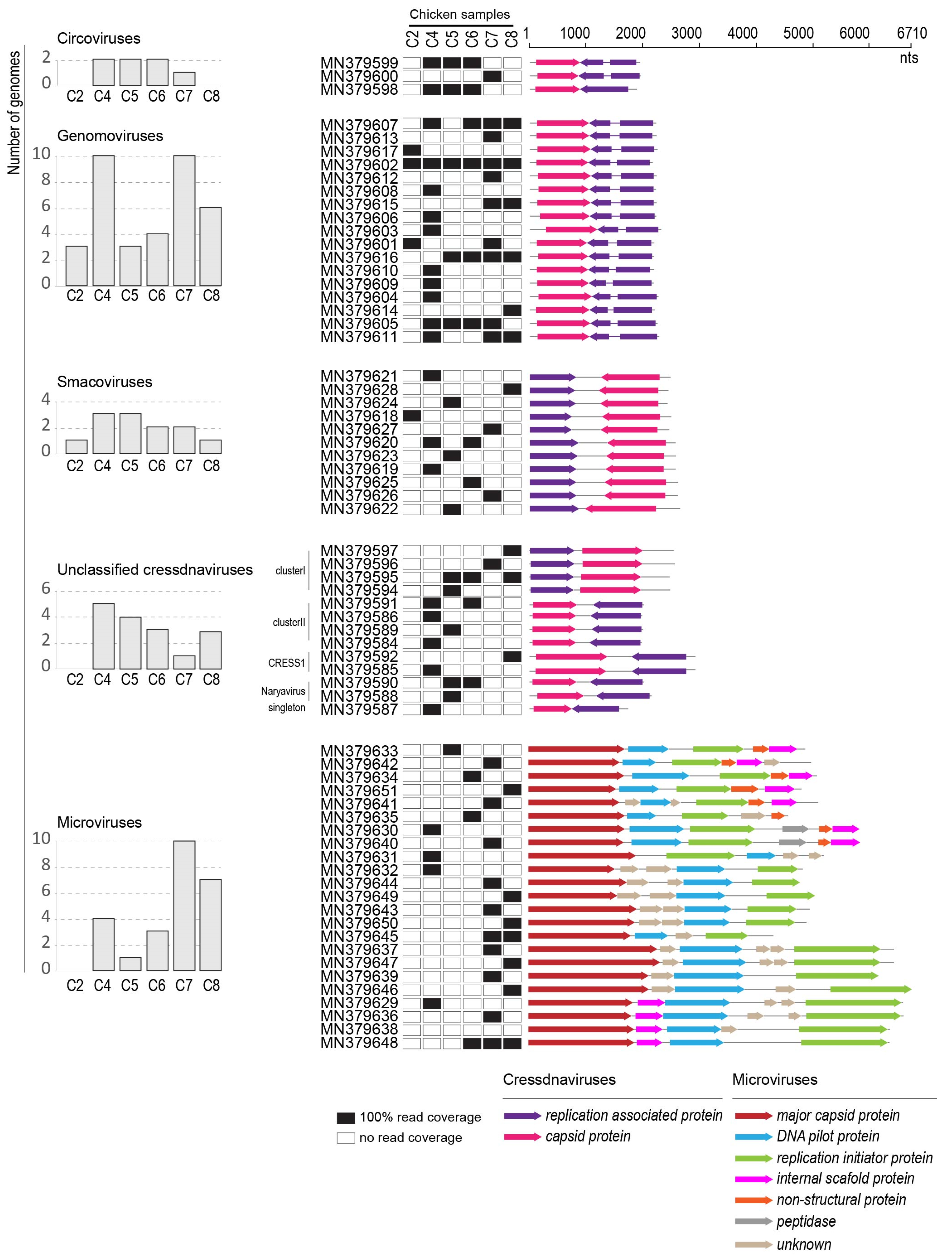
3.1. Identification of Circular Single Stranded DNA Viruses in Swab Samples
3.2. Cressdnaviruses
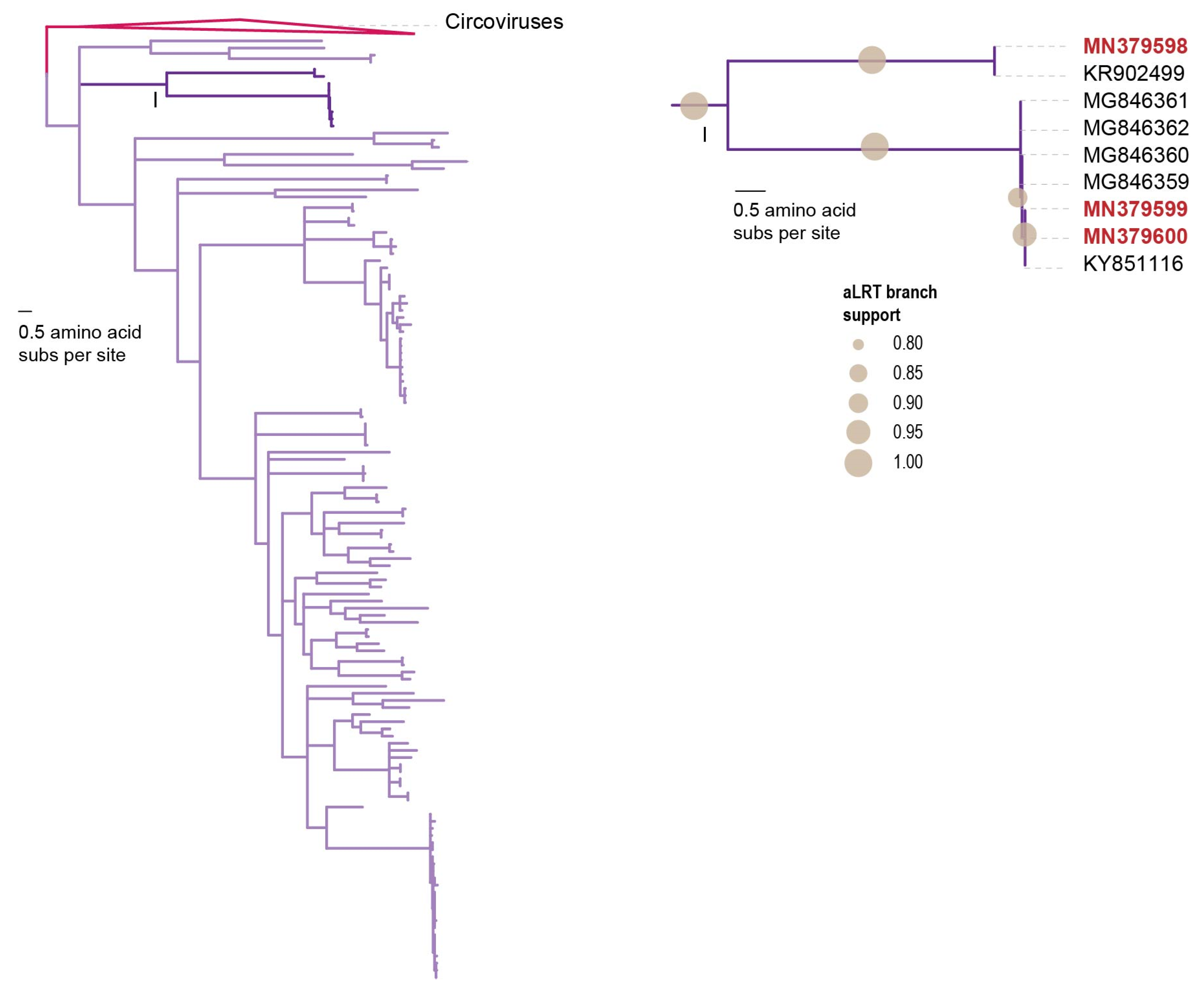
3.2.1. Circoviruses
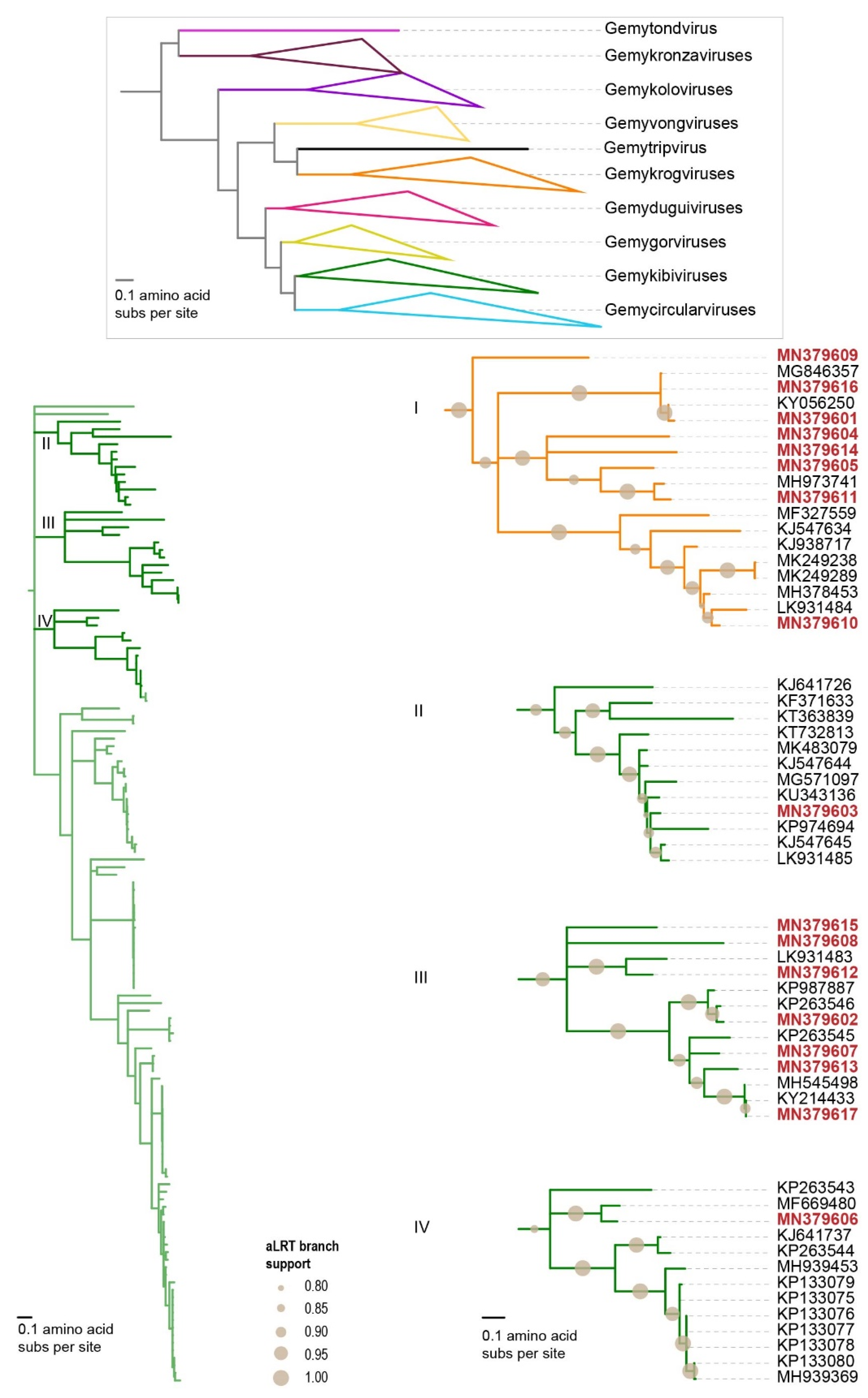
3.2.2. Genomoviruses
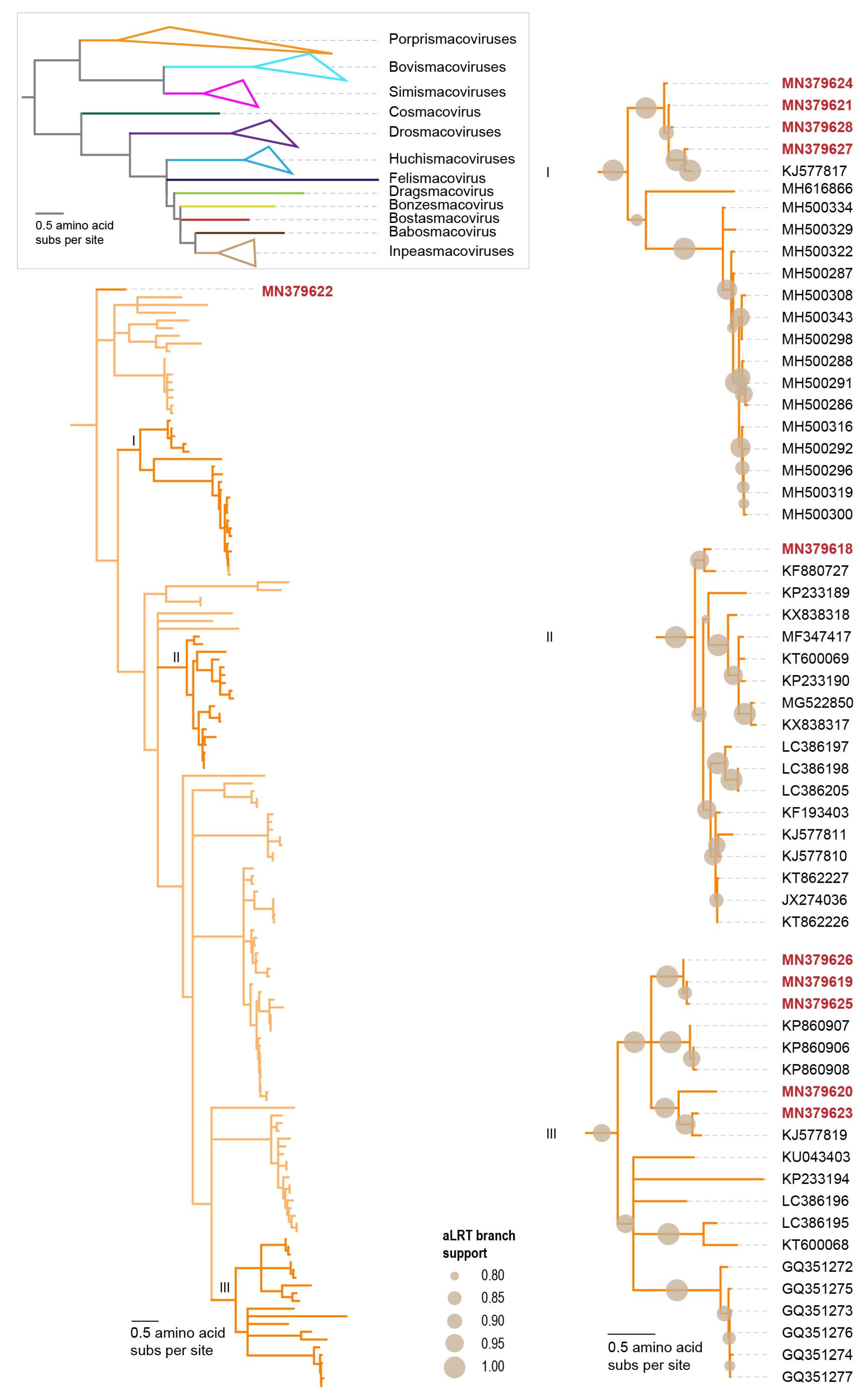
3.2.3. Smacoviruses
3.2.4. Unclassified Cressdnaviruses
3.3. Microviruses
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Nayfach, S.; Paez-Espino, D.; Call, L.; Low, S.J.; Sberro, H.; Ivanova, N.N.; Proal, A.D.; Fischbach, M.A.; Bhatt, A.S.; Hugenholtz, P.; et al. Metagenomic compendium of 189,680 DNA viruses from the human gut microbiome. Nat. Microbiol. 2021, 6, 960–970. [Google Scholar] [CrossRef] [PubMed]
- Tisza, M.J.; Buck, C.B. A catalog of tens of thousands of viruses from human metagenomes reveals hidden associations with chronic diseases. Proc. Natl. Acad. Sci. USA 2021, 118, e2023202118. [Google Scholar] [CrossRef]
- Tisza, M.J.; Pastrana, D.V.; Welch, N.L.; Stewart, B.; Peretti, A.; Starrett, G.J.; Pang, Y.S.; Krishnamurthy, S.R.; Pesavento, P.A.; McDermott, D.H.; et al. Discovery of several thousand highly diverse circular DNA viruses. Elife 2020, 9, 555375. [Google Scholar] [CrossRef]
- Asplund, M.; Kjartansdottir, K.R.; Mollerup, S.; Vinner, L.; Fridholm, H.; Herrera, J.A.R.; Friis-Nielsen, J.; Hansen, T.A.; Jensen, R.H.; Nielsen, I.B.; et al. Contaminating viral sequences in high-throughput sequencing viromics: A linkage study of 700 sequencing libraries. Clin. Microbiol. Infect. 2019, 25, 1277–1285. [Google Scholar] [CrossRef] [Green Version]
- Holmes, E.C. Reagent contamination in viromics: All that glitters is not gold. Clin. Microbiol. Infect. 2019, 25, 1167–1168. [Google Scholar] [CrossRef] [Green Version]
- Porter, A.F.; Cobbin, J.; Li, C.; Eden, J.-S.; Holmes, E.C. Metagenomic identification of viral sequences in laboratory reagents. Viruses 2021, 13, 2122. [Google Scholar] [CrossRef]
- Krupovic, M.; Varsani, A.; Kazlauskas, D.; Breitbart, M.; Delwart, E.; Rosario, K.; Yutin, N.; Wolf, Y.I.; Harrach, B.; Zerbini, F.M.; et al. Cressdnaviricota: A Virus Phylum Unifying Seven Families of Rep-Encoding Viruses with Single-Stranded, Circular DNA Genomes. J. Virol. 2020, 94, e00582-20. [Google Scholar] [CrossRef]
- Kraberger, S.; Schmidlin, K.; Fontenele, R.S.; Walters, M.; Varsani, A. Unravelling the Single-Stranded DNA Virome of the New Zealand Blackfly. Viruses 2019, 11, 532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de la Higuera, I.; Kasun, G.W.; Torrance, E.L.; Pratt, A.A.; Maluenda, A.; Colombet, J.; Bisseux, M.; Ravet, V.; Dayaram, A.; Stainton, D.; et al. Unveiling Crucivirus Diversity by Mining Metagenomic Data. Mbio 2020, 11, e01410–e01420. [Google Scholar] [CrossRef]
- Levy, H.; Fontenele, R.S.; Harding, C.; Suazo, C.; Kraberger, S.; Schmidlin, K.; Djurhuus, A.; Black, C.E.; Hart, T.; Smith, A.L.; et al. Identification and Distribution of Novel Cressdnaviruses and Circular molecules in Four Penguin Species in South Georgia and the Antarctic Peninsula. Viruses 2020, 12, 1029. [Google Scholar] [CrossRef] [PubMed]
- Kinsella, C.M.; Bart, A.; Deijs, M.; Broekhuizen, P.; Kaczorowska, J.; Jebbink, M.F.; van Gool, T.; Cotten, M.; van der Hoek, L. Entamoeba and Giardia parasites implicated as hosts of CRESS viruses. Nat. Commun. 2020, 11, 4620. [Google Scholar] [CrossRef]
- Custer, J.M.; White, R.; Taylor, H.; Schmidlin, K.; Fontenele, R.S.; Stainton, D.; Kraberger, S.; Briskie, J.V.; Varsani, A. Diverse single-stranded DNA viruses identified in New Zealand (Aotearoa) South Island robin (Petroica australis) fecal samples. Virology 2021, 565, 38–51. [Google Scholar] [CrossRef] [PubMed]
- Cherwa, J.E.J.; Fane, B.A. Microviridae. In Virus Taxonomy; King, A.M.Q., Adams, M.J., Carstens, E.B., Lefkowitz, E.J., Eds.; Elsevier: San Diego, CA, USA, 2012; pp. 385–393. [Google Scholar]
- Breitbart, M.; Fane, B.A. Microviridae. eLS 2021, 1–14. [Google Scholar] [CrossRef]
- Collins, C.L.; DeNardo, D.F.; Blake, M.; Norton, J.; Schmidlin, K.; Fontenele, R.S.; Wilson, M.A.; Kraberger, S.; Varsani, A. Genome Sequences of Microviruses Identified in Gila Monster Feces. Microbiol. Resour. Announc. 2021, 10, e00163-21. [Google Scholar] [CrossRef] [PubMed]
- Kraberger, S.; Cook, C.N.; Schmidlin, K.; Fontenele, R.S.; Bautista, J.; Smith, B.; Varsani, A. Diverse single-stranded DNA viruses associated with honey bees (Apis mellifera). Infect. Genet. Evol. 2019, 71, 179–188. [Google Scholar] [CrossRef] [PubMed]
- Labonte, J.M.; Suttle, C.A. Metagenomic and whole-genome analysis reveals new lineages of gokushoviruses and biogeographic separation in the sea. Front. Microbiol. 2013, 4, 404. [Google Scholar] [CrossRef]
- Roux, S.; Krupovic, M.; Poulet, A.; Debroas, D.; Enault, F. Evolution and diversity of the Microviridae viral family through a collection of 81 new complete genomes assembled from virome reads. PLoS ONE 2012, 7, e40418. [Google Scholar] [CrossRef] [Green Version]
- Creasy, A.; Rosario, K.; Leigh, B.A.; Dishaw, L.J.; Breitbart, M. Unprecedented Diversity of ssDNA Phages from the Family Microviridae Detected within the Gut of a Protochordate Model Organism (Ciona robusta). Viruses 2018, 10, 404. [Google Scholar] [CrossRef] [Green Version]
- Kirchberger, P.C.; Ochman, H. Resurrection of a global, metagenomically defined gokushovirus. Elife 2020, 9, e51599. [Google Scholar] [CrossRef] [PubMed]
- Swayne, D.E.; Pantin-Jackwood, M. Pathogenicity of avian influenza viruses in poultry. Dev. Biol. 2006, 124, 61–67. [Google Scholar]
- Alexander, D.J. Newcastle disease and other avian paramyxoviruses. Rev. Sci. Et Tech. -Off. Int. Des Epizoot. 2000, 19, 443–462. [Google Scholar] [CrossRef] [PubMed]
- Tulman, E.; Afonso, C.; Lu, Z.; Zsak, L.; Rock, D.; Kutish, G. The genome of a very virulent Marek’s disease virus. J. Virol. 2000, 74, 7980–7988. [Google Scholar] [CrossRef] [Green Version]
- Zhao, K.; He, W.; Xie, S.; Song, D.; Lu, H.; Pan, W.; Zhou, P.; Liu, W.; Lu, R.; Zhou, J.; et al. Highly pathogenic fowlpox virus in cutaneously infected chickens, China. Emerg. Infect. Dis. 2014, 20, 1208–1210. [Google Scholar] [CrossRef] [PubMed]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. 2010. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 15 February 2021).
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [Green Version]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Muhire, B.M.; Varsani, A.; Martin, D.P. SDT: A virus classification tool based on pairwise sequence alignment and identity calculation. PLoS ONE 2014, 9, e108277. [Google Scholar] [CrossRef]
- Bushnell, B. BBMap Short-Read Aligner, and Other Bioinformatics Tools. 2015. Available online: https://jgi.doe.gov/data-and-tools/bbtools/ (accessed on 15 February 2021).
- Zallot, R.; Oberg, N.; Gerlt, J.A. The EFI Web Resource for Genomic Enzymology Tools: Leveraging Protein, Genome, and Metagenome Databases to Discover Novel Enzymes and Metabolic Pathways. Biochemistry 2019, 58, 4169–4182. [Google Scholar] [CrossRef]
- Orton, J.P.; Morales, M.; Fontenele, R.S.; Schmidlin, K.; Kraberger, S.; Leavitt, D.J.; Webster, T.H.; Wilson, M.A.; Kusumi, K.; Dolby, G.A.; et al. Virus Discovery in Desert Tortoise Fecal Samples: Novel Circular Single-Stranded DNA Viruses. Viruses 2020, 12, v12020143. [Google Scholar] [CrossRef] [Green Version]
- Fontenele, R.S.; Lacorte, C.; Lamas, N.S.; Schmidlin, K.; Varsani, A.; Ribeiro, S.G. Single Stranded DNA Viruses Associated with Capybara Faeces Sampled in Brazil. Viruses 2019, 11, 710. [Google Scholar] [CrossRef] [Green Version]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Walker, P.J.; Siddell, S.G.; Lefkowitz, E.J.; Mushegian, A.R.; Adriaenssens, E.M.; Alfenas-Zerbini, P.; Davison, A.J.; Dempsey, D.M.; Dutilh, B.E.; Garcia, M.L.; et al. Changes to virus taxonomy and to the International Code of Virus Classification and Nomenclature ratified by the International Committee on Taxonomy of Viruses (2021). Arch. Virol. 2021, 166, 2633–2648. [Google Scholar] [CrossRef] [PubMed]
- Varsani, A.; Martin, D.P.; Randles, J.W.; Vetten, H.J.; Thomas, J.E.; Fiallo-Olive, E.; Navas-Castillo, J.; Lett, J.M.; Zerbini, F.M.; Roumagnac, P.; et al. Taxonomy update for the family Alphasatellitidae: New subfamily, genera, and species. Arch. Virol. 2021, 166, 3503–3511. [Google Scholar] [CrossRef]
- Gronenborn, B.; Randles, J.W.; Knierim, D.; Barriere, Q.; Vetten, H.J.; Warthmann, N.; Cornu, D.; Sileye, T.; Winter, S.; Timchenko, T. Analysis of DNAs associated with coconut foliar decay disease implicates a unique single-stranded DNA virus representing a new taxon. Sci. Rep. 2018, 8, 5698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kazlauskas, D.; Varsani, A.; Koonin, E.V.; Krupovic, M. Multiple origins of prokaryotic and eukaryotic single-stranded DNA viruses from bacterial and archaeal plasmids. Nat. Commun. 2019, 10, 3425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kazlauskas, D.; Varsani, A.; Krupovic, M. Pervasive Chimerism in the Replication-Associated Proteins of Uncultured Single-Stranded DNA Viruses. Viruses 2018, 10, 187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Capella-Gutierrez, S.; Silla-Martinez, J.M.; Gabaldon, T. trimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef] [PubMed]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef] [Green Version]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef] [PubMed]
- Guindon, S.; Dufayard, J.F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [Green Version]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. ProtTest 3: Fast selection of best-fit models of protein evolution. Bioinformatics 2011, 27, 1164–1165. [Google Scholar] [CrossRef] [Green Version]
- Stover, B.C.; Muller, K.F. TreeGraph 2: Combining and visualizing evidence from different phylogenetic analyses. BMC Bioinformatics 2010, 11, 7. [Google Scholar] [CrossRef] [Green Version]
- Pei, J.; Kim, B.H.; Grishin, N.V. PROMALS3D: A tool for multiple protein sequence and structure alignments. Nucleic Acids Res. 2008, 36, 2295–2300. [Google Scholar] [CrossRef] [PubMed]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2--approximately maximum-likelihood trees for large alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef] [PubMed]
- Thomas, J.E.; Gronenborn, B.; Harding, R.M.; Mandal, B.; Grigoras, I.; Randles, J.W.; Sano, Y.; Timchenko, T.; Vetten, H.J.; Yeh, H.H.; et al. ICTV Virus Taxonomy Profile: Nanoviridae. J. Gen. Virol. 2021, 102, 001544. [Google Scholar] [CrossRef]
- Male, M.F.; Kraberger, S.; Stainton, D.; Kami, V.; Varsani, A. Cycloviruses, gemycircularviruses and other novel replication-associated protein encoding circular viruses in Pacific flying fox (Pteropus tonganus) faeces. Infect. Genet. Evol. 2016, 39, 279–292. [Google Scholar] [CrossRef]
- Rosario, K.; Duffy, S.; Breitbart, M. A field guide to eukaryotic circular single-stranded DNA viruses: Insights gained from metagenomics. Arch. Virol. 2012, 157, 1851–1871. [Google Scholar] [CrossRef] [PubMed]
- Nash, T.E.; Dallas, M.B.; Reyes, M.I.; Buhrman, G.K.; Ascencio-Ibanez, J.T.; Hanley-Bowdoin, L. Functional analysis of a novel motif conserved across geminivirus Rep proteins. J. Virol. 2011, 85, 1182–1192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kraberger, S.; Waits, K.; Ivan, J.; Newkirk, E.; VandeWoude, S.; Varsani, A. Identification of circular single-stranded DNA viruses in faecal samples of Canada lynx (Lynx canadensis), moose (Alces alces) and snowshoe hare (Lepus americanus) inhabiting the Colorado San Juan Mountains. Infect. Genet. Evol. 2018, 64, 1–8. [Google Scholar] [CrossRef]
- Breitbart, M.; Delwart, E.; Rosario, K.; Segales, J.; Varsani, A.; Ictv Report, C. ICTV Virus Taxonomy Profile: Circoviridae. J. Gen. Virol. 2017, 98, 1997–1998. [Google Scholar] [CrossRef]
- Rosario, K.; Breitbart, M.; Harrach, B.; Segales, J.; Delwart, E.; Biagini, P.; Varsani, A. Revisiting the taxonomy of the family Circoviridae: Establishment of the genus Cyclovirus and removal of the genus Gyrovirus. Arch. Virol. 2017, 162, 1447–1463. [Google Scholar] [CrossRef] [Green Version]
- Victoria, J.G.; Kapoor, A.; Li, L.; Blinkova, O.; Slikas, B.; Wang, C.; Naeem, A.; Zaidi, S.; Delwart, E. Metagenomic analyses of viruses in stool samples from children with acute flaccid paralysis. J. Virol. 2009, 83, 4642–4651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phan, T.G.; Luchsinger, V.; Avendano, L.F.; Deng, X.; Delwart, E. Cyclovirus in nasopharyngeal aspirates of Chilean children with respiratory infections. J. Gen. Virol. 2014, 95, 922–927. [Google Scholar] [CrossRef]
- Tan, L.V.; van Doorn, H.R.; Nghia, H.D.; Chau, T.T.; Tu, L.T.P.; de Vries, M.; Canuti, M.; Deijs, M.; Jebbink, M.F.; Baker, S.; et al. Identification of a new cyclovirus in cerebrospinal fluid of patients with acute central nervous system infections. MBio 2013, 4, e00231-00213. [Google Scholar] [CrossRef] [Green Version]
- Yan, T.; Li, G.; Zhou, D.; Yang, X.; Hu, L.; Cheng, Z. Novel Cyclovirus Identified in Broiler Chickens With Transmissible Viral Proventriculitis in China. Front. Vet. Sci. 2020, 7, 569098. [Google Scholar] [CrossRef]
- Lima, D.A.; Cibulski, S.P.; Tochetto, C.; Varela, A.P.M.; Finkler, F.; Teixeira, T.F.; Loiko, M.R.; Cerva, C.; Junqueira, D.M.; Mayer, F.Q. The intestinal virome of malabsorption syndrome-affected and unaffected broilers through shotgun metagenomics. Virus Res. 2019, 261, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Kapoor, A.; Slikas, B.; Bamidele, O.S.; Wang, C.; Shaukat, S.; Masroor, M.A.; Wilson, M.L.; Ndjango, J.B.; Peeters, M.; et al. Multiple diverse circoviruses infect farm animals and are commonly found in human and chimpanzee feces. J. Virol. 2010, 84, 1674–1682. [Google Scholar] [CrossRef] [Green Version]
- Feher, E.; Kaszab, E.; Forro, B.; Bali, K.; Marton, S.; Lengyel, G.; Banyai, K. Genome sequence of a mallard duck origin cyclovirus, DuACyV-1. Arch. Virol. 2017, 162, 3925–3929. [Google Scholar] [CrossRef]
- Li, L.; Giannitti, F.; Low, J.; Keyes, C.; Ullmann, L.S.; Deng, X.; Aleman, M.; Pesavento, P.A.; Pusterla, N.; Delwart, E. Exploring the virome of diseased horses. J. Gen. Virol. 2015, 96, 2721–2733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varsani, A.; Krupovic, M. Family Genomoviridae: 2021 taxonomy update. Arch. Virol. 2021, 166, 2911–2926. [Google Scholar] [CrossRef]
- Yu, X.; Li, B.; Fu, Y.; Jiang, D.; Ghabrial, S.A.; Li, G.; Peng, Y.; Xie, J.; Cheng, J.; Huang, J.; et al. A geminivirus-related DNA mycovirus that confers hypovirulence to a plant pathogenic fungus. Proc. Natl. Acad. Sci. USA 2010, 107, 8387–8392. [Google Scholar] [CrossRef] [Green Version]
- Li, P.; Wang, S.; Zhang, L.; Qiu, D.; Zhou, X.; Guo, L. A tripartite ssDNA mycovirus from a plant pathogenic fungus is infectious as cloned DNA and purified virions. Sci. Adv. 2020, 6, eaay9634. [Google Scholar] [CrossRef] [Green Version]
- Steel, O.; Kraberger, S.; Sikorski, A.; Young, L.M.; Catchpole, R.J.; Stevens, A.J.; Ladley, J.J.; Coray, D.S.; Stainton, D.; Dayaram, A.; et al. Circular replication-associated protein encoding DNA viruses identified in the faecal matter of various animals in New Zealand. Infect. Genet. Evol. 2016, 43, 151–164. [Google Scholar] [CrossRef]
- Lima, D.A.; Cibulski, S.P.; Finkler, F.; Teixeira, T.F.; Varela, A.P.M.; Cerva, C.; Loiko, M.R.; Scheffer, C.M.; Dos Santos, H.F.; Mayer, F.Q.; et al. Faecal virome of healthy chickens reveals a large diversity of the eukaryote viral community, including novel circular ssDNA viruses. J. Gen. Virol. 2017, 98, 690–703. [Google Scholar] [CrossRef]
- Rosario, K.; Mettel, K.A.; Benner, B.E.; Johnson, R.; Scott, C.; Yusseff-Vanegas, S.Z.; Baker, C.C.; Cassill, D.L.; Storer, C.; Varsani, A. Virus discovery in all three major lineages of terrestrial arthropods highlights the diversity of single-stranded DNA viruses associated with invertebrates. PeerJ 2018, 6, e5761. [Google Scholar] [CrossRef]
- Krupovic, M.; Varsani, A. A 2021 taxonomy update for the family Smacoviridae. Arch. Virol. 2021, 166, 3245–3253. [Google Scholar] [CrossRef]
- Diez-Villasenor, C.; Rodriguez-Valera, F. CRISPR analysis suggests that small circular single-stranded DNA smacoviruses infect Archaea instead of humans. Nat. Commun. 2019, 10, 294. [Google Scholar] [CrossRef] [Green Version]
- Varsani, A.; Krupovic, M. Smacoviridae: A new family of animal-associated single-stranded DNA viruses. Arch. Virol. 2018, 163, 2005–2015. [Google Scholar] [CrossRef] [PubMed]
- Cibulski, S.; Alves de Lima, D.; Fernandes Dos Santos, H.; Teixeira, T.F.; Tochetto, C.; Mayer, F.Q.; Roehe, P.M. A plate of viruses: Viral metagenomics of supermarket chicken, pork and beef from Brazil. Virology 2021, 552, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Dayaram, A.; Potter, K.A.; Pailes, R.; Marinov, M.; Rosenstein, D.D.; Varsani, A. Identification of diverse circular single-stranded DNA viruses in adult dragonflies and damselflies (Insecta: Odonata) of Arizona and Oklahoma, USA. Infect. Genet. Evol. 2015, 30, 278–287. [Google Scholar] [CrossRef] [PubMed]
- Tochetto, C.; Muterle Varela, A.P.; Alves de Lima, D.; Loiko, M.R.; Mengue Scheffer, C.; Pinto Paim, W.; Cerva, C.; Schmidt, C.; Cibulski, S.P.; Cano Ortiz, L.; et al. Viral DNA genomes in sera of farrowing sows with or without stillbirths. PLoS ONE 2020, 15, e0230714. [Google Scholar] [CrossRef]
- Khalifeh, A.; Blumstein, D.T.; Fontenele, R.S.; Schmidlin, K.; Richet, C.; Kraberger, S.; Varsani, A. Diverse cressdnaviruses and an anellovirus identified in the fecal samples of yellow-bellied marmots. Virology 2021, 554, 89–96. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genome Type | Family | Genera |
|---|---|---|
| dsDNA | Adenoviridae | Atadenovirus, Aviadenovirus, Mastadenovirus, Siadenovirus |
| Herpesviridae | Iltovirus, Mardivirus | |
| Poxviridae | Avipoxvirus | |
| ssDNA | Anelloviridae | Gyrovirus |
| Circoviridae | Cyclovirus | |
| Genomoviridae | Gemycircularvirus, Gemykibivirus | |
| Parvoviridae | Aveparvovirus, Dependoparvovirus, Protoparvovirus | |
| Smacoviridae | Huchismacovirus | |
| dsRNA | Birnaviridae | Avibirnavirus |
| Picobirnaviridae | Picobirnavirus | |
| Reoviridae | Orthoreovirus, Rotavirus | |
| ssRNA (+) | Astroviridae | Avastrovirus |
| Caliciviridae | Bavovirus, Norovirus | |
| Coronaviridae | Deltacoronavirus, Gammacoronavirus | |
| Flaviviridae | Flavivirus | |
| Hepeviridae | Orthohepevirus | |
| Picornaviridae | Aphthovirus, Avisivirus, Enterovirus, Gallivirus, Megrivirus, Orivirus, Sicinivirus, Tremovirus | |
| ssRNA (-) | Orthomyxoviridae | Alphainfluenzavirus |
| Paramyxoviridae | Avulavirus, Orthoavulavirus, Paraavulavirus | |
| Peribunyaviridae | Orthobunyavirus | |
| Pneumoviridae | Metapneumovirus | |
| Rhabdoviridae | Lyssavirus, Sunrhavirus | |
| RNA-RT | Retroviridae | Alpharetrovirus, Gammaretrovirus |
| Contigs in Each Sample | ||||||||
|---|---|---|---|---|---|---|---|---|
| Family | Length of Contigs (nt) | C2 | C4 | C5 | C6 | C7 | C8 | Total Contigs |
| Ackermannviridae | 762–8399 | - | - | 1 | 1 | - | - | 2 |
| Anelloviridae | 815 | - | - | - | - | 1 | - | 1 |
| Autographiviridae | 1170–1440 | 1 | - | 2 | - | - | - | 3 |
| Circoviridae | 771–2329 | - | 2 | 3 | 7 | 2 | 3 | 17 |
| Demerecviridae | 772–2046 | - | 1 | - | - | 1 | 2 | 4 |
| Genomoviridae | 760–2347 | 5 | 11 | 4 | 4 | 11 | 8 | 43 |
| Herelleviridae | 751–6913 | 1 | - | - | 1 | 3 | 9 | 14 |
| Inoviridae | 867–5794 | 2 | - | - | - | - | 2 | 4 |
| Microviridae | 781–6787 | 1 | 25 | 16 | 24 | 24 | 36 | 126 |
| Myoviridae | 754–8471 | 23 | 3 | 17 | 11 | 9 | 56 | 119 |
| Parvoviridae | 785–2429 | - | 2 | - | - | - | - | 2 |
| Podoviridae | 769–14,239 | 11 | 1 | 3 | 9 | 9 | 10 | 43 |
| Siphoviridae | 751–11,004 | 32 | 6 | 23 | 24 | 10 | 58 | 153 |
| Smacoviridae | 896–2385 | 3 | 3 | 7 | 4 | 4 | 4 | 25 |
| unclassified cressdnaviruses | 768–5193 | - | 25 | 20 | 24 | 15 | 25 | 109 |
| Family | Genus/Group | Species | Accession | Name | Motif I | Motif II | GRS | Motif III | Walker A | Walker B | Motif C |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Circoviridae | Cyclovirus | Duck-associated cyclovirus 1 | MN379599 | Chicken cyclovirus mg5_2967 | SWTLNN | PHLQG | - | QNHDYCSK | GASGTGKSRRAA | IIDDF | ITSN |
| MN379600 | Chicken cyclovirus mg7_102 | SWTLNN | PHLQG | - | QNHDYCAK | GASGTGKSRRAA | IIDDF | ITSN | |||
| Horse-associated cyclovirus 1 | MN379598 | Chicken cyclovirus mg4_1122 | CFTYNN | KHLQG | - | QNYDYCTK | GETGTGKSRKCA | IIDDF | ITSN | ||
| Genomoviridae | Gemykibivirus | Gemykibivirus anima1 | MN379613 | Chicken genomovirus mg7_74 | LFTYSQ | THLHV | RKFDVEDFHPNIVPSL | GGWDYATK | GRSRTGKTILAR | VFDDI | WIMN |
| MN379617 | Chicken genomovirus mg2_274u | LFTYSQ | THLHV | RKFDVEGFHPNIVPSL | GGWDYATK | GRSRTGKTWLAR | VFDDI | WIMN | |||
| Gemykibivirus cowchi1 | MN379606 | Chicken genomovirus mg4_1196 | LLTYAQ | THLHS | DAFDVGGYHPNISPSY | KGFDYTIK | GPSRLGKTLWAR | VFDDI | WLSN | ||
| Gemykibivirus galga1 | MN379612 | Chicken genomovirus mg7_73 | LLTYAQ | THLHA | SVFDVAGFHPNISITK | IHYDYAIK | GKSRTGKTNYAR | VFDDI | WISN | ||
| Gemykibivirus galga2 | MN379615 | Chicken genomovirus mg8_401 | LLTYSQ | THLHA | DVYDVDGFHPNISPSL | RGYDYAIK | GPTRTGKTMWSR | VFDDV | WLAN | ||
| Gemykibivirus galga3 | MN379608 | Chicken genomovirus mg4_1218 | LLTYPQ | NHLHA | DVFDVDGRHPNIQSRL | AGYDYVIK | GDTLTGKTQWAR | IFDDL | YISN | ||
| Gemykibivirus humas1 | MN379603 | Chicken genomovirus mg4_1107 | LLTYPQ | IHLHA | RAFDVEGCHPNVSPSR | DGYDYAIK | GPSRMGKTIWAR | IFDDF | WLSN | ||
| Gemykibivirus humas3 | MN379602 | Chicken genomovirus mg2_77 | LFTYSQ | THLHA | RKFDVEGFHPNIISTI | GSWDYATK | GPSRTGKTMWAR | VFDDI | WLSN | ||
| Gemykibivirus monas1 | MN379607 | Chicken genomovirus mg4_1210 | LFTYSQ | SHLHV | RKFDVEGFHPNIVPSL | GGWDYATK | GPSRTGKTMWAR | VFDDI | WLMN | ||
| Gemykrogvirus | Gemykrogvirus apime1 | MN379611 | Chicken genomovirus mg7_70 | FLTYSQ | HHFHA | SRLDFGCHHPNIQSVR | RTWDYVGK | GPTRTGKTANIL | VFDDI | MLMN | |
| Gemykrogvirus carib1 | MN379610 | Chicken genomovirus mg4_1259 | IITFPQ | VHYHV | TAFDYFGAHGNIKSVR | KVFDYVGK | GPTRTGKTLYAR | VFDDI | MCMN | ||
| Gemykrogvirus galga1 | MN379609 | Chicken genomovirus mg4_1247 | LLTYSQ | THFHV | RLFDFGSSHPNIQIIR | KAFDYAGK | GPTRSGKSVWPR | VFDDL | MCMN | ||
| Gemykrogvirus galga2 | MN379601 | Chicken genomovirus mg2_75 | FLTYSQ | SHLHC | SLFDYRGAHPNIKSIR | KPWNYAGK | GPSRTGKTVWAR | IFDDI | MCMN | ||
| MN379616 | Chicken genomovirus mg8_416 | FLTYSQ | SHLHC | SLFDYRGAHPNIKSIR | KPWNYAGK | GPSRTGKTVWAR | IFDDI | MCMN | |||
| Gemykrogvirus galga3 | MN379605 | Chicken genomovirus mg4_1173 | FLTYSQ | CHFHV | SRLDFGGHHPNIQSVR | RVWDYAGK | GPTRTGKTVWAR | VFDDI | MLMN | ||
| Gemykrogvirus galga4 | MN379614 | Chicken genomovirus mg7_78 | LLTYSK | VHVHC | FRFDFGGSHPNIRSVS | RTYDYAGK | GPTRTGKTVWAR | VFDDL | CLMN | ||
| Gemykrogvirus galga5 | MN379604 | Chicken genomovirus mg4_1165 | LLTYSQ | LHFHC | SRLDYNGSHPNIKPIR | RAWEYTGK | GESRTGKTIWAR | IFDDI | LLCN | ||
| Smacoviridae | Porprismacovirus | Porprismacovirus chicas2 | MN379619 | Chicken smacovirus mg4_881 | MITMPR | QHWQC | - | DTWEYETK | PEGNHGKTWLVG | FIDIP | VMTN |
| MN379625 | Chicken smacovirus mg6_1052 | MITMPR | QHWQC | - | DTWDYETK | PEGNHGKTWLVG | FIDIP | VMTN | |||
| MN379626 | Chicken smacovirus mg7_57 | MITMPR | QHWQC | - | DTWDYETK | PEGNHGKTWLVG | FIDIP | VMTN | |||
| Porprismacovirus chicas3 | MN379623 | Chicken smacovirus mg5_1212 | MLTIPR | EHWQV | - | DKWEYETK | PKGNNGKSWLVG | VIDLP | VLTN | ||
| Porprismacovirus chicas4 | MN379620 | Chicken smacovirus mg4_885 | MLTIPR | KHWQV | - | EDSDYETK | PKGKAGKSWLIG | IIDMP | VLTN | ||
| Porprismacovirus chicas5 | MN379627 | Chicken smacovirus mg7_67 | MLTIPR | DHWQI | - | DKWEYERK | PIGNRGKSWLAG | VIDIP | ILTN | ||
| Porprismacovirus chicas6 | MN379621 | Chicken smacovirus mg4_964 | MLTIPR | EHWQV | - | DKWEYERK | PIGNRGKSWLAG | VIDIP | IMTN | ||
| MN379624 | Chicken smacovirus mg5_1444 | MMTIPR | DHWQV | - | DKWEYEKK | PIGNRGKSWLAG | VIDIP | IMTN | |||
| MN379628 | Chicken smacovirus mg8_345 | MLTIPR | EHWQI | - | DRWEYERK | PIGNRGKSWLAG | VIDIP | IMTN | |||
| Porprismacovirus chicas7 | MN379622 | Chicken smacovirus mg5_1081 | MLTIPQ | KHWQI | - | DVWDYERK | KSGNHGKTWLSQ | IIDIP | IFTN | ||
| Porprismacovirus turas1 | MN379618 | Chicken smacovirus mg2_55 | IMTIPQ | EHWQI | - | DECLYERK | AGGNVGKSWFCG | IIDVP | CLTN | ||
| unclassified | ClusterI | MN379594 | Chicken virus mg5_1345 | DATIWI | RHYQF | - | RDFEYVYK | EVGNAGKTAWGM | IIDTP | VLCN | |
| ClusterI | MN379595 | Chicken virus mg6_1197 | DATIWC | RHFQF | - | RDFEYVYK | ERGNSGKTAWAM | IIDTP | ILCN | ||
| ClusterI | MN379596 | Chicken virus mg7_59 | DATIWC | RHFQF | - | RDFEYVYK | ERGNSGKTAWAM | IIDTP | VLCN | ||
| ClusterI | MN379597 | Chicken virus mg8_324 | DATIWC | RHFQF | - | RDFEYVYK | ERGNSGKTAWAM | IIDTP | VLCN | ||
| ClusterII | MN379584 | Chicken virus mg4_280 | QLTLNQ | EHIHI | - | QNIDYIRK | GAPGVGKTYSAY | VVEEF | FASV | ||
| ClusterII | MN379591 | Chicken virus mg6_2056 | QITLNE | KHIHI | - | QNIAYIEK | GDSGSGKTYKAY | VIEEF | IASI | ||
| ClusterII | MN379586 | Chicken virus mg4_1578 | QITLND | KHIHI | - | QNIDYIEK | GDSGTGKTYKAY | IVEEF | IASI | ||
| ClusterII | MN379589 | Chicken virus mg5_2676 | QLTLNQ | EHIHI | - | QNIEYIRK | GAPGVGKTYSAY | VVEEF | FASV | ||
| CRESS1 | MN379585 | Chicken virus mg4_657 | CFTSFK | KHLQG | - | KAIEYCKK | GQAGSGKSHHCY | WFDEF | ISTT | ||
| CRESS1 | MN379592 | Chicken virus mg8_273 | CFTSFK | KHLQG | - | KAIEYCKK | GQAGSGKSHHCY | WFDEF | ISTT | ||
| Naryavirus | MN379588 | Chicken virus mg5_2197 | QITLNE | EHIHC | - | QNVAYVKK | GPSGVGKTERAK | IYDDF | ITSV | ||
| Naryavirus | MN379590 | Chicken virus mg5_2876 | QLTLNE | EHIHI | - | QNIDYILK | GPSGVGKTNKAL | LYDDF | ITSV | ||
| Singleton | MN379587 | Chicken virus mg4_2302 | IFTINN | QHLQG | - | QAYNYATK | GDSGTGKSYSAR | VLDEF | ITTN |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chrzastek, K.; Kraberger, S.; Schmidlin, K.; Fontenele, R.S.; Kulkarni, A.; Chappell, L.; Dufour-Zavala, L.; Kapczynski, D.R.; Varsani, A. Diverse Single-Stranded DNA Viruses Identified in Chicken Buccal Swabs. Microorganisms 2021, 9, 2602. https://doi.org/10.3390/microorganisms9122602
Chrzastek K, Kraberger S, Schmidlin K, Fontenele RS, Kulkarni A, Chappell L, Dufour-Zavala L, Kapczynski DR, Varsani A. Diverse Single-Stranded DNA Viruses Identified in Chicken Buccal Swabs. Microorganisms. 2021; 9(12):2602. https://doi.org/10.3390/microorganisms9122602
Chicago/Turabian StyleChrzastek, Klaudia, Simona Kraberger, Kara Schmidlin, Rafaela S. Fontenele, Arun Kulkarni, Len Chappell, Louise Dufour-Zavala, Darrell R. Kapczynski, and Arvind Varsani. 2021. "Diverse Single-Stranded DNA Viruses Identified in Chicken Buccal Swabs" Microorganisms 9, no. 12: 2602. https://doi.org/10.3390/microorganisms9122602