
Microbial Community Dynamics during Biodegradation of Crude Oil and Its Response to Biostimulation in Svalbard Seawater at Low Temperature




Abstract
:1. Introduction
2. Materials and Methods
2.1. Experimental Setup and Sampling
2.2. Chemical Analyses
2.3. DNA Extraction
2.4. Quantitative PCR Conditions and Data Analysis
2.5. Database of Genera Containing Oil Hydrocarbon-Degrading Organisms
2.6. Taxonomic Profiling of Prokaryotic Community
2.6.1. Shotgun Metagenomic Sequencing
2.6.2. Amplicon-Based Sequencing
2.6.3. Comparison and Integration of Bacterial Community Taxonomic Classification Methods
3. Results
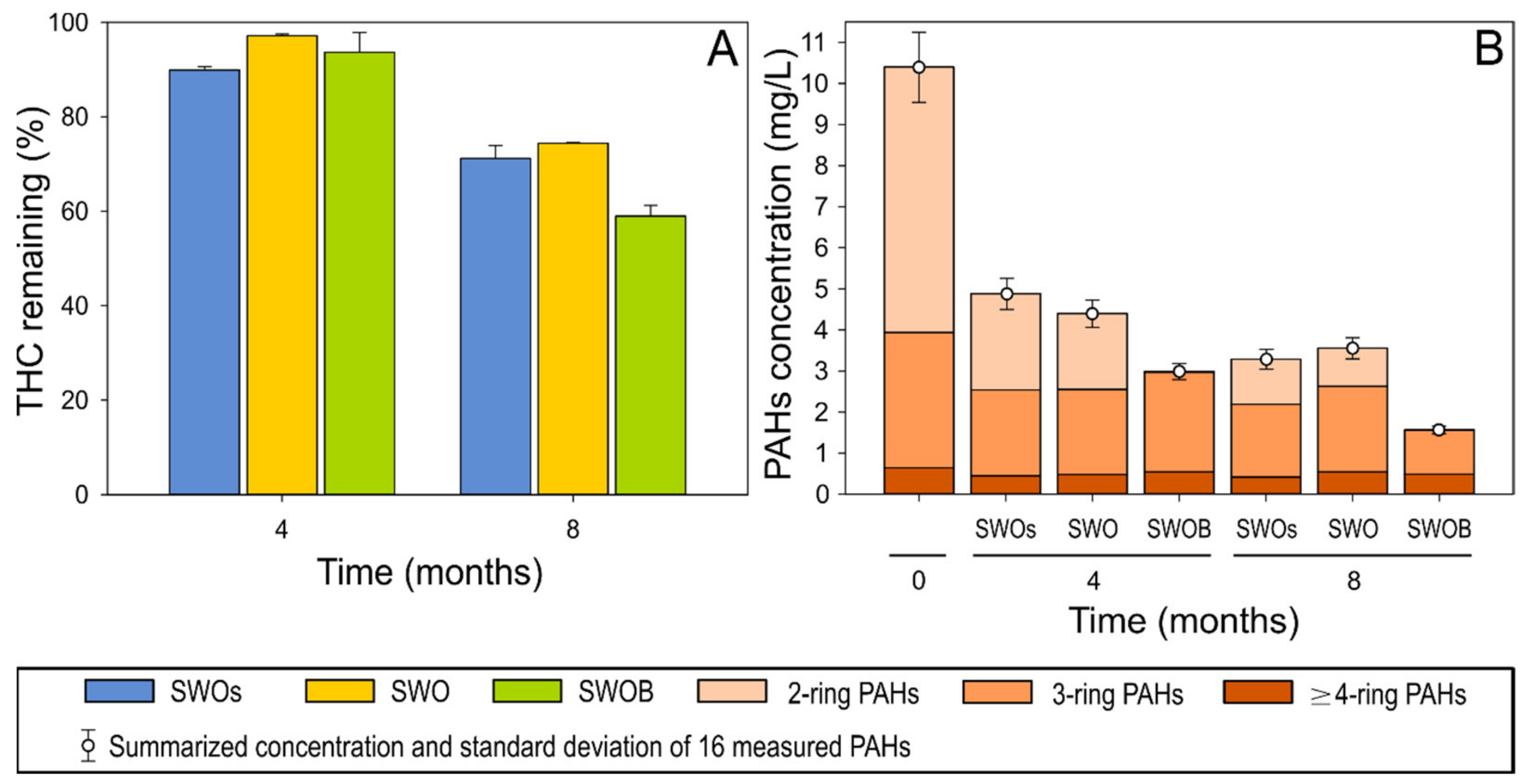
3.1. Oil Hydrocarbon Depletion
3.2. Microbial Community Abundance and Composition
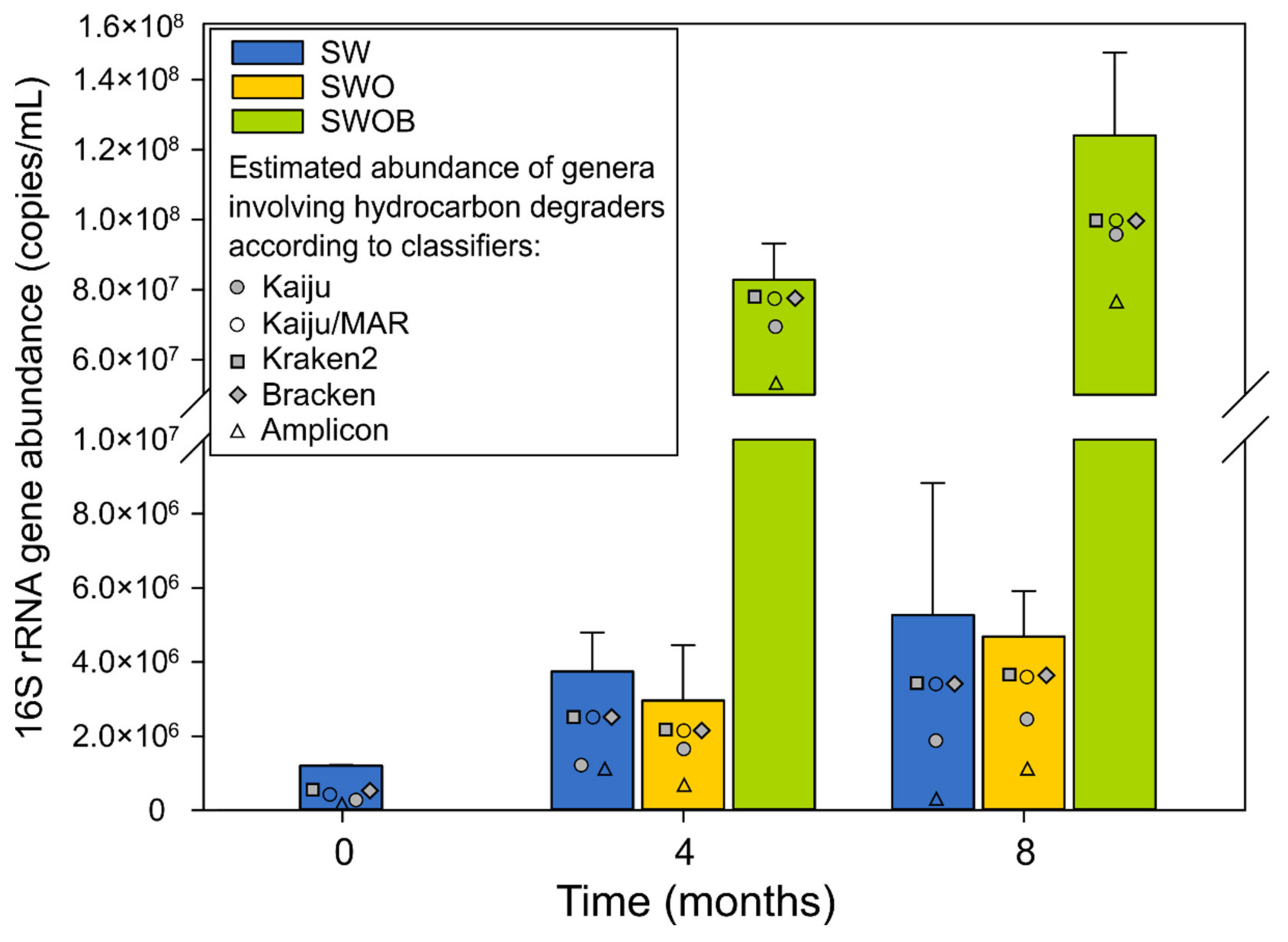
3.2.1. Microbial Community Abundance
3.2.2. Microbial Community Structure
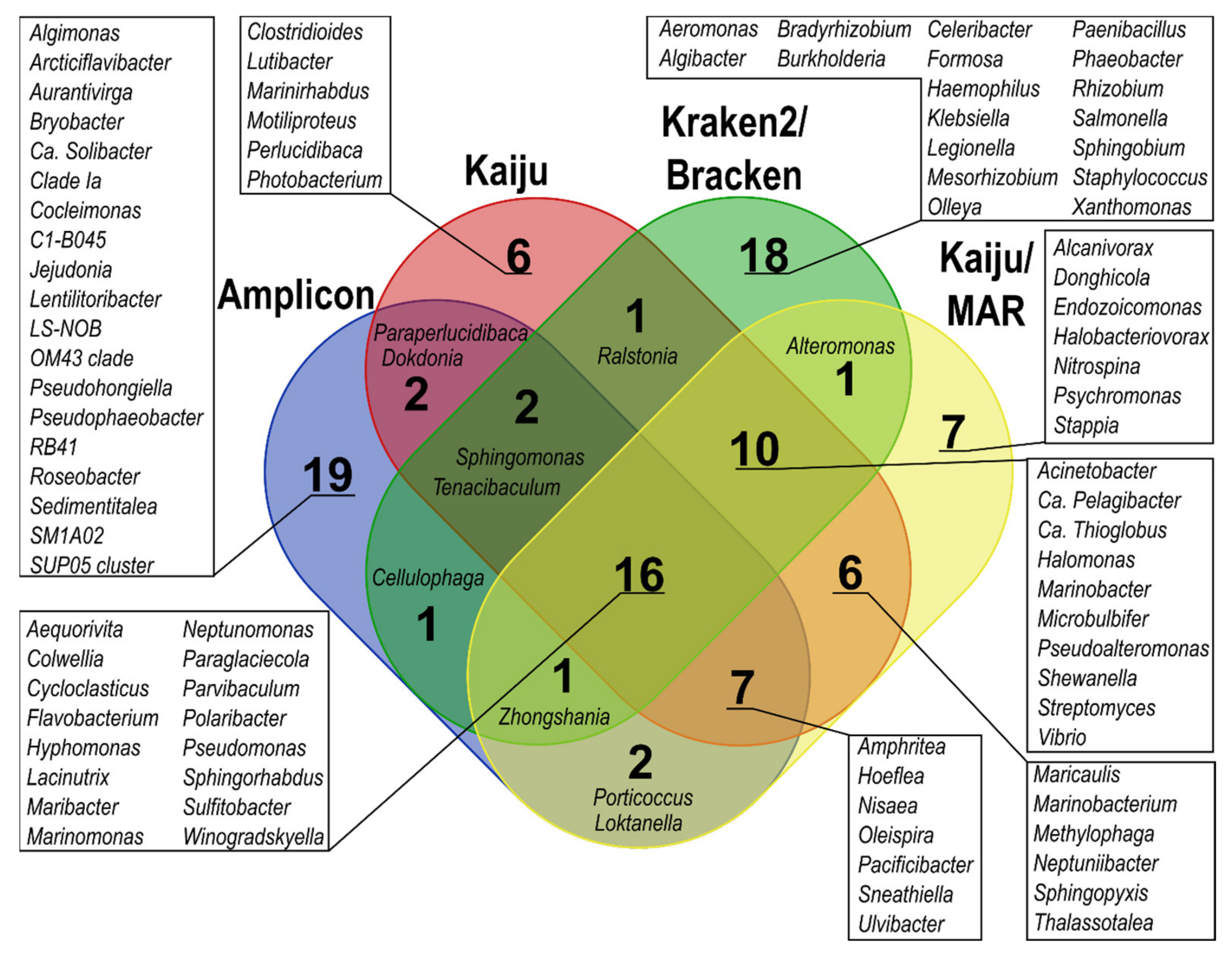
Estimation of Bacterial Community Structure According to Different Classification Methods
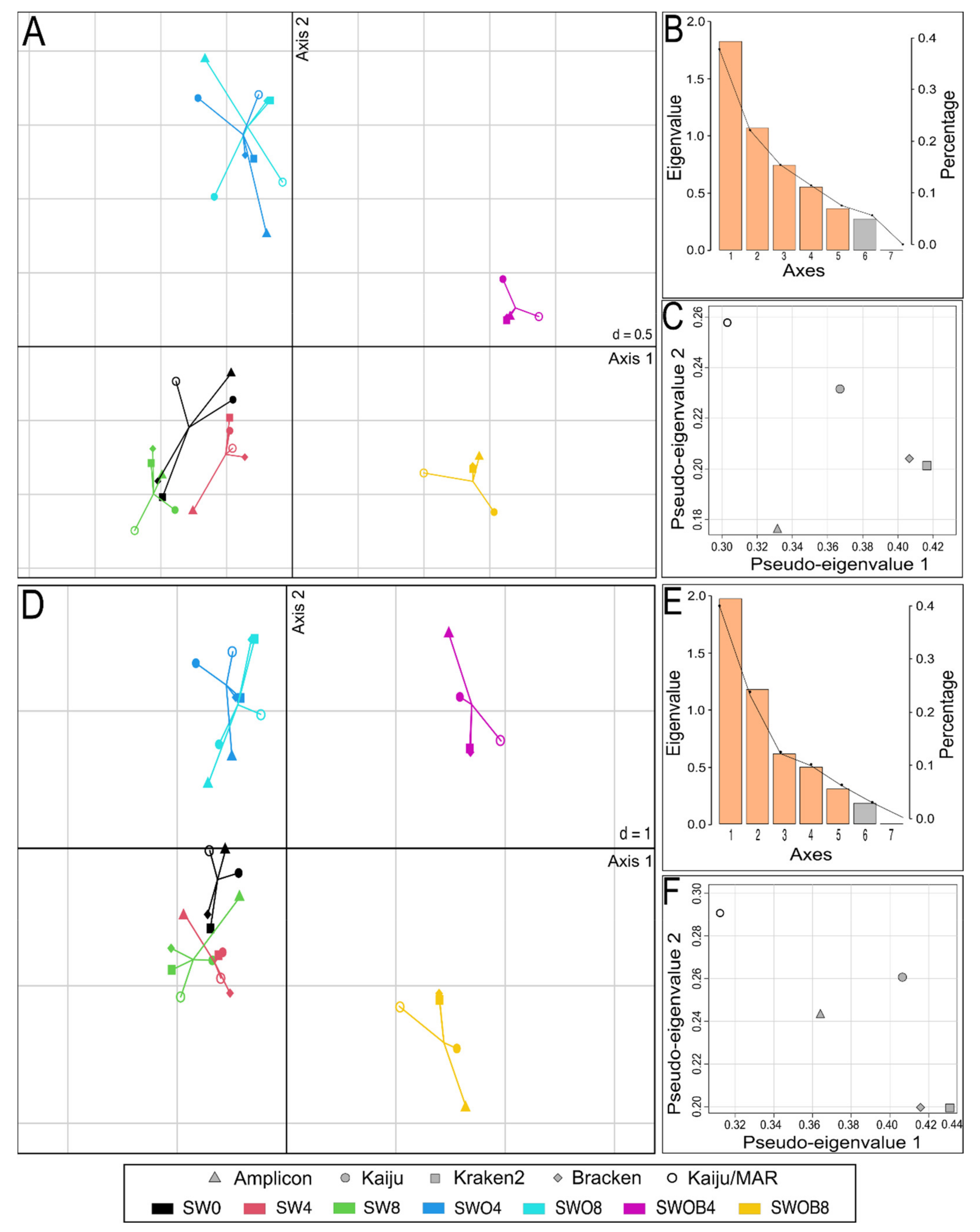
Estimation of Bacterial Community Similarity According to Different Classification Methods
Estimation of Bacterial Genera Proportions via Quantification
3.3. Hydrocarbon Degradation Potential of Bacterial Community
3.3.1. The Dynamics of Estimated Abundances of Genera Containing Hydrocarbon Degraders
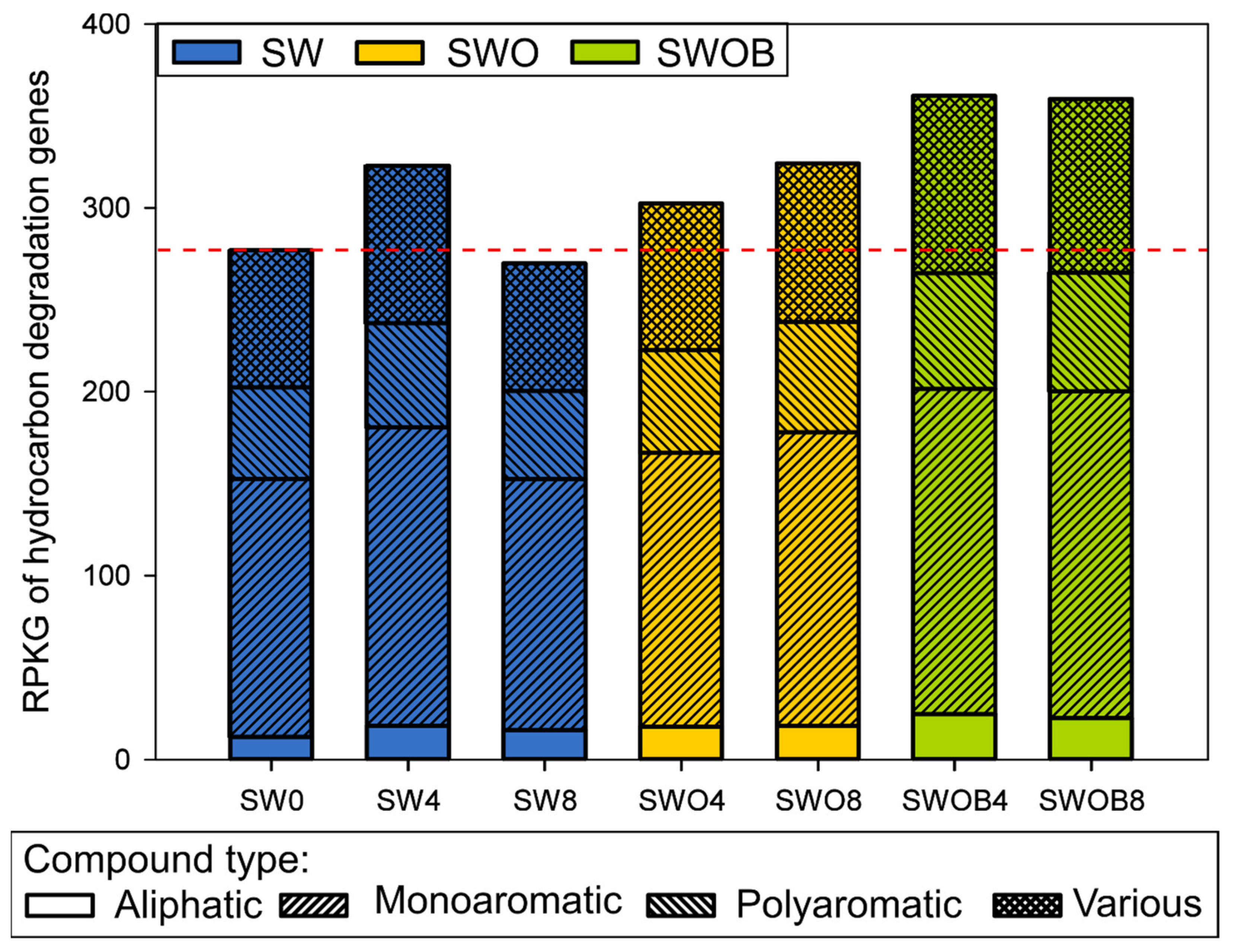
3.3.2. The Dynamics of Hydrocarbon Degradation-Related Genes
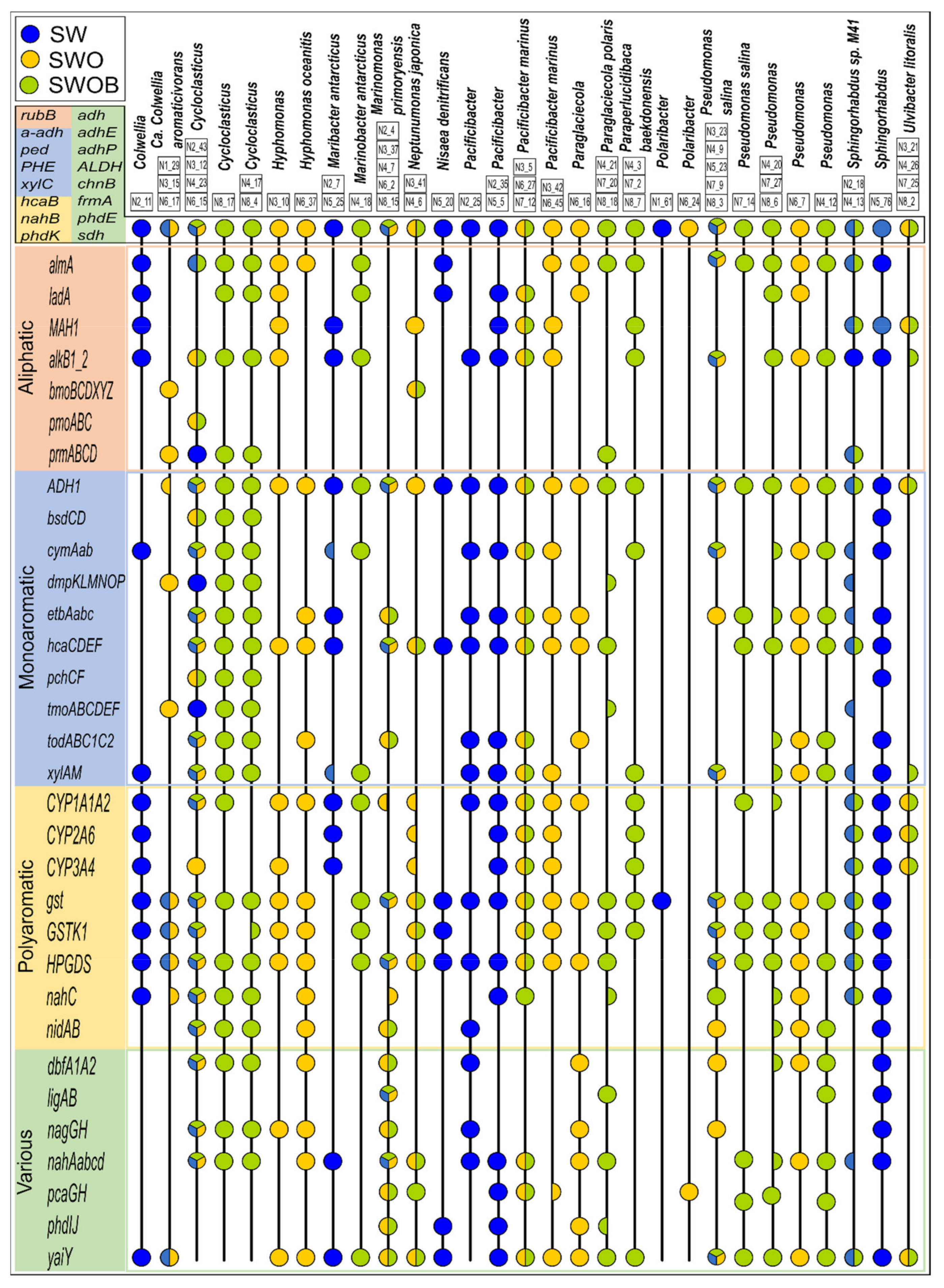
3.3.3. MAGs and Their Oil Hydrocarbon Degradation Potential
4. Discussion
4.1. The Effect of Taxonomic Classification Method on the Estimation of Community Composition in Arctic Seawater-Derived Bacterial Communities
4.2. Bacterial Community Potential for Oil Hydrocarbons Degradation in Arctic Seawater
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Boylan, B.M. Increased maritime traffic in the Arctic: Implications for governance of Arctic Sea routes. Mar. Policy 2021, 131, 104566. [Google Scholar] [CrossRef]
- Ranellone, R.T.; Tukaew, P.; Shi, X.; Rangwala, A.S. Ignitability of crude oil and its oil-in-water products at arctic temperature. Mar. Pollut. Bull. 2017, 115, 261–265. [Google Scholar] [CrossRef] [PubMed]
- Hazen, T.C.; Prince, R.C.; Mahmoudi, N. Marine Oil Biodegradation. Environ. Sci. Technol. 2016, 50, 2121–2129. [Google Scholar] [CrossRef] [PubMed]
- Brakstad, O.G.; Lofthus, S.; Ribicic, D.; Netzer, R. Biodegradation of petroleum oil in cold marine environments. In Psychrophiles: From Biodiversity to Biotechnology, 2nd ed.; Springer International Publishing: New York, NY, USA, 2017; pp. 613–644. [Google Scholar] [CrossRef]
- Nikolopoulou, M.; Pasadakis, N.; Kalogerakis, N. Evaluation of autochthonous bioaugmentation and biostimulation during microcosm-simulated oil spills. Mar. Pollut. Bull. 2013, 72, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Crisafi, F.; Genovese, M.; Smedile, F.; Russo, D.; Catalfamo, M.; Yakimov, M.; Giuliano, L.; Denaro, R. Bioremediation technologies for polluted seawater sampled after an oil-spill in Taranto Gulf (Italy): A comparison of biostimulation, bioaugmentation and use of a washing agent in microcosm studies. Mar. Pollut. Bull. 2016, 106, 119–126. [Google Scholar] [CrossRef] [PubMed]
- Hassanshahian, M.; Bayat, Z.; Cappello, S.; Smedile, F.; Yakimov, M. Comparison the effects of bioaugmentation versus biostimulation on marine microbial community by PCR-DGGE: A mesocosm scale. J. Environ. Sci. 2016, 43, 136–146. [Google Scholar] [CrossRef]
- Al-Mailem, D.M.; Al-Deieg, M.; Eliyas, M.; Radwan, S.S. Biostimulation of indigenous microorganisms for bioremediation of oily hypersaline microcosms from the Arabian Gulf Kuwaiti coasts. J. Environ. Manag. 2017, 193, 576–583. [Google Scholar] [CrossRef]
- Brakstad, O.G.; Throne-Holst, M.; Netzer, R.; Stoeckel, D.M.; Atlas, R.M. Microbial communities related to biodegradation of dispersed Macondo oil at low seawater temperature with Norwegian coastal seawater. Microb. Biotechnol. 2015, 8, 989–998. [Google Scholar] [CrossRef]
- Brakstad, O.G.; Davies, E.J.; Ribicic, D.; Winkler, A.; Brönner, U.; Netzer, R. Biodegradation of dispersed oil in natural seawaters from Western Greenland and a Norwegian fjord. Polar Biol. 2018, 41, 2435–2450. [Google Scholar] [CrossRef] [Green Version]
- Ribicic, D.; Netzer, R.; Hazen, T.C.; Techtmann, S.M.; Drabløs, F.; Brakstad, O.G. Microbial community and metagenome dynamics during biodegradation of dispersed oil reveals potential key-players in cold Norwegian seawater. Mar. Pollut. Bull. 2018, 129, 370–378. [Google Scholar] [CrossRef] [Green Version]
- Tovo, A.; Menzel, P.; Krogh, A.; Lagomarsino, M.C.; Suweis, S. Taxonomic classification method for metagenomics based on core protein families with Core-Kaiju. Nucleic Acids Res. 2020, 48, e93. [Google Scholar] [CrossRef]
- Vergeynst, L.; Christensen, J.H.; Kjeldsen, K.U.; Meire, L.; Boone, W.; Malmquist, L.M.V.; Rysgaard, S. In situ biodegradation, photooxidation and dissolution of petroleum compounds in Arctic seawater and sea ice. Water Res. 2019, 148, 459–468. [Google Scholar] [CrossRef] [Green Version]
- Yergeau, E.; Michel, C.; Tremblay, J.; Niemi, A.; King, T.L.; Wyglinski, J.; Lee, K.; Greer, C.W. Metagenomic survey of the taxonomic and functional microbial communities of seawater and sea ice from the Canadian Arctic. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [Green Version]
- Cao, S.; Zhang, W.; Ding, W.; Wang, M.; Fan, S.; Yang, B.; Mcminn, A.; Wang, M.; Xie, B.; Qin, Q.-L.; et al. Structure and function of the Arctic and Antarctic marine microbiota as revealed by metagenomics. Microbiome 2020, 8, 47. [Google Scholar] [CrossRef] [Green Version]
- Ruijter, J.M.; Ramakers, C.; Hoogaars, W.M.H.; Karlen, Y.; Bakker, O.; van den Hoff, M.J.B.; Moorman, A.F.M. Amplification efficiency: Linking baseline and bias in the analysis of quantitative PCR data. Nucleic Acids Res. 2009, 37, e45. [Google Scholar] [CrossRef] [Green Version]
- Nõlvak, H.; Truu, M.; Kanger, K.; Tampere, M.; Espenberg, M.; Loit, E.; Raave, H.; Truu, J. Inorganic and organic fertilizers impact the abundance and proportion of antibiotic resistance and integron-integrase genes in agricultural grassland soil. Sci. Total Environ. 2016, 562, 678–689. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc (accessed on 10 October 2021).
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011, 17, 10. [Google Scholar] [CrossRef]
- Rodriguez, L.M.; Gunturu, S.; Tiedje, J.M.; Cole, J.R.; Konstantinidis, K.T. Nonpareil 3: Fast estimation of metagenomic coverage and sequence diversity. mSystems 2018, 3, e00039. [Google Scholar] [CrossRef] [Green Version]
- Menzel, P.; Ng, K.L.; Krogh, A. Fast and sensitive taxonomic classification for metagenomics with Kaiju. Nat. Commun. 2016, 7, 11257. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Luo, R.; Liu, C.-M.; Leung, C.-M.; Ting, H.-F.; Sadakane, K.; Yamashita, H.; Lam, T.W. MEGAHIT W1.0: A fast and scalable metagenome assembler driven by advanced methodologies and community practices. Methods 2016, 102, 3–11. [Google Scholar] [CrossRef]
- Kang, D.D.; Li, F.; Kirton, E.; Thomas, A.; Egan, R.; An, H.; Wang, Z. MetaBAT 2: An adaptive binning algorithm for robust and efficient genome reconstruction from metagenome assemblies. PeerJ 2019, 7, e7359. [Google Scholar] [CrossRef]
- Parks, D.H.; Imelfort, M.; Skennerton, C.T.; Hugenholtz, P.; Tyson, G.W. CheckM: Assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015, 25, 1043–1055. [Google Scholar] [CrossRef] [Green Version]
- Olm, M.R.; Brown, C.T.; Brooks, B.; Banfield, J.F. DRep: A tool for fast and accurate genomic comparisons that enables improved genome recovery from metagenomes through de-replication. ISME J. 2017, 11, 2864–2868. [Google Scholar] [CrossRef] [Green Version]
- Kanehisa, M.; Goto, S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Wang, W.; Shao, Z. Diversity of flavin-binding monooxygenase genes (almA) in marine bacteria capable of degradation long-chain alkanes. FEMS Microbiol. Ecol. 2012, 80, 523–533. [Google Scholar] [CrossRef] [Green Version]
- Nayfach, S.; Pollard, K.S. Average genome size estimation improves comparative metagenomics and sheds light on the functional ecology of the human microbiome. Genome Biol. 2015, 16, 51. [Google Scholar] [CrossRef] [Green Version]
- Parada, A.E.; Needham, D.M.; Fuhrman, J.A. Every base matters: Assessing small subunit rRNA primers for marine microbiomes with mock communities, time series and global field samples. Environ. Microbiol. 2016, 18, 1403–1414. [Google Scholar] [CrossRef]
- Parameswaran, P.; Jalili, R.; Tao, L.; Shokralla, S.; Gharizadeh, B.; Ronaghi, M.; Fire, A.Z. A pyrosequencing-tailored nucleotide barcode design unveils opportunities for large-scale sample multiplexing. Nucleic Acids Res. 2007, 35, e130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herbold, C.W.; Pelikan, C.; Kuzyk, O.; Hausmann, B.; Angel, R.; Berry, D.; Loy, A. A flexible and economical barcoding approach for highly multiplexed amplicon sequencing of diverse target genes. Front. Microbiol. 2015, 6, 731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Kobert, K.; Flouri, T.; Stamatakis, A. PEAR: A fast and accurate Illumina Paired-End reAd mergeR. Bioinformatics 2014, 30, 614–620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J.; et al. Introducing mothur: Open-Source, Platform-Independent, Community-Supported Software for Describing and Comparing Microbial Communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef] [Green Version]
- Rognes, T.; Flouri, T.; Nichols, B.; Quince, C.; Mahé, F. VSEARCH: A versatile open source tool for metagenomics. PeerJ 2016, 4, e2584. [Google Scholar] [CrossRef]
- Westcott, S.L.; Schloss, P.D. De novo clustering methods outperform reference-based methods for assigning 16S rRNA gene sequences to operational taxonomic units. PeerJ 2015, 12, e1487. [Google Scholar] [CrossRef]
- Klemetsen, T.; Raknes, I.A.; Fu, J.; Agafonov, A.; Balasundaram, S.V.; Tartari, G.; Robertsen, E.; Willassen, N.P. The MAR databases: Development and implementation of databases specific for marine metagenomics. Nucleic Acids Res. 2018, 46, D693–D699. [Google Scholar] [CrossRef]
- Wood, D.E.; Salzberg, S.L. Kraken: Ultrafast metagenomic sequence classification using exact alignments. Genome Biol. 2014, 15. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.; Breitwieser, F.P.; Thielen, P.; Salzberg, S.L. Bracken: Estimating species abundance in metagenomics data. PeerJ Comput. Sci. 2017, 3, e104. [Google Scholar] [CrossRef]
- Pruesse, E.; Quast, C.; Knittel, K.; Fuchs, B.M.; Ludwig, W.; Peplies, J.; Glöckner, F.O. SILVA: A comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res. 2007, 35, 7188–7196. [Google Scholar] [CrossRef] [Green Version]
- Metsalu, T.; Vilo, J. ClustVis: A web tool for visualizing clustering of multivariate data using Principal Component Analysis and heatmap. Nucleic Acids Res. 2015, 43, W566–W570. [Google Scholar] [CrossRef]
- Meng, C.; Kuster, B.; Culhane, A.C.; Gholami, A.M. A multivariate approach to the integration of multi-omics datasets. BMC Bioinform. 2014, 15, 162. [Google Scholar] [CrossRef] [Green Version]
- Fadeev, E.; Cardozo-Mina, M.G.; Rapp, J.Z.; Bienhold, C.; Salter, I.; Salman-Carvalho, V.; Molari, M.; Tegetmeyer, H.E.; Buttigieg, P.L.; Boetius, A. Comparison of two 16S rRNA primers (V3–V4 and V4–V5) for studies of Arctic microbial communities. Front. Microbiol. 2021, 12, 283. [Google Scholar] [CrossRef]
- Ye, S.H.; Siddle, K.J.; Park, D.J.; Sabeti, P.C. Benchmarking Metagenomics Tools for Taxonomic Classification. Cell 2019, 178, 779–794. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Huang, S.; Zhang, M.; Zhu, Q.; Haiminen, N.; Carrieri, A.P.; Vázquez-Baeza, Y.; Parida, L.; Kim, H.-C.; Knight, R.; et al. Challenges in benchmarking metagenomic profilers. Nat. Methods 2021, 18, 618–626. [Google Scholar] [CrossRef] [PubMed]
- Tessler, M.; Neumann, J.S.; Afshinnekoo, E.; Pineda, M.; Hersch, R.; Velho, L.F.M.; Segovia, B.T.; Lansac-Toha, F.A.; Lemke, M.; DeSalle, R.; et al. Large-scale differences in microbial diversity discovery between 16S amplicon and shotgun sequencing. Sci. Rep. 2017, 7, 6589. [Google Scholar] [CrossRef] [PubMed]
- Logares, R.; Sunagawa, S.; Salazar, G.; Cornejo-Castillo, F.M.; Ferrera, I.; Sarmento, H.; Hingamp, P.; Ogata, H.; de Vargas, C.; Lima-Mendez, G.; et al. Metagenomic 16S rDNA Illumina tags are a powerful alternative to amplicon sequencing to explore diversity and structure of microbial communities. Environ. Microbiol. 2014, 16, 2659–2671. [Google Scholar] [CrossRef]
- Wood, D.E.; Lu, J.; Langmead, B. Improved metagenomic analysis with Kraken 2. Genome Biol. 2019, 20, 257. [Google Scholar] [CrossRef] [Green Version]
- Nasko, D.J.; Koren, S.; Phillippy, A.M.; Treangen, T.J. RefSeq database growth influences the accuracy of k-mer-based lowest common ancestor species identification. Genome Biol. 2018, 19, 165. [Google Scholar] [CrossRef] [Green Version]
- Saati-Santamaría, Z.; Peral-Aranega, E.; Velázquez, E.; Rivas, R.; García-Fraile, P. Phylogenomic analyses of the genus Pseudomonas lead to the rearrangement of several species and the definition of new genera. Biology 2021, 10, 782. [Google Scholar] [CrossRef]
- Brakstad, O.G.; Nonstad, I.; Faksness, L.-G.; Brandvik, P.J. Responses of microbial communities in arctic sea ice after contamination by crude petroleum oil. Microb. Ecol. 2008, 55, 540–552. [Google Scholar] [CrossRef]
- Nie, Y.; Chi, C.-Q.; Fang, H.; Liang, S.-L.; Lai, G.-L.; Tang, Y.-Q.; Wu, X.-L. Diverse alkane hydroxylase genes in microorganisms and environments. Sci. Rep. 2014, 4, 4968. [Google Scholar] [CrossRef] [Green Version]
- Redmond, M.C.; Valentine, D.L. Natural gas and temperature structured a microbial community response to the Deepwater Horizon oil spill. Proc. Nat. Acad. Sci. USA 2012, 109, 20292–20297. [Google Scholar] [CrossRef] [Green Version]
- Gutierrez, T.; Singleton, D.R.; Berry, D.; Yang, T.; Aitken, M.D.; Teske, A. Hydrocarbon-degrading bacteria enriched by the Deepwater Horizon oil spill identified by cultivation and DNA-SIP. ISME J. 2013, 7, 2091–2104. [Google Scholar] [CrossRef]
- Dong, C.; Bai, X.; Lai, Q.; Xie, Y.; Chen, X.; Shao, Z. Draft genome sequence of Marinomonas sp. Strain D104, a Polycyclic aromatic hydrocarbon-degrading bacterium from the deep-sea sediment of the Arctic ocean. Genome Announc. 2015, 2, e1211-13. [Google Scholar] [CrossRef] [Green Version]
- Crisafi, F.; Giuliano, L.; Yakimov, M.M.; Azzaro, M.; Denaro, R. Isolation and degradation potential of a cold-adapted oil/PAH-degrading marine bacterial consortium from Kongsfjorden (Arctic region). Rend. Fis. Acc. Lincei 2016, 27, S261–S270. [Google Scholar] [CrossRef]
- McFarlin, K.M.; Prince, R.C.; Perkins, R.; Leigh, M.B. Biodegradation of dispersed oil in Arctic seawater at −1 degrees C. PLoS ONE 2014, 9, e84297. [Google Scholar] [CrossRef] [Green Version]
- Kristensen, M.; Johnsen, A.R.; Christensen, J.H. Marine biodegradation of crude oil in temperate and Arctic water samples. J. Hazard. Mater. 2015, 300, 75–83. [Google Scholar] [CrossRef]
- Scheibye, K.; Christensen, J.H.; Johnsen, A.R. Biodegradation of crude oil in Arctic subsurface water from the Disko Bay (Greenland) is limited. Environ. Pollut. 2017, 223, 73–80. [Google Scholar] [CrossRef]
- Yang, S.; Wen, X.; Shi, Y.; Liebner, S.; Jin, H.; Perfumo, A. Hydrocarbon degraders establish at the costs of microbial richness, abundance and keystone taxa after crude oil contamination in permafrost environments. Sci. Rep. 2016, 6, 37473. [Google Scholar] [CrossRef]
- Prince, R.C. Oil spill dispersants: Boon or bane? Environ. Sci. Technol. 2015, 49, 6376–6384. [Google Scholar] [CrossRef] [Green Version]
- Yakimov, M.M.; Giuliano, L.; Gentile, G.; Crisafi, E.; Chernikova, T.N.; Abraham, W.R.; Lünsdorf, H.; Timmis, K.N.; Golyshin, P.N. Oleispira antarctica gen. nov., sp. nov., a new hydrocarbonoclastic marine bacterium, isolated from an Antarctic coastal seawater. Int. J. Syst. Evolut. Microbiol. 2003, 53, 779–785. [Google Scholar] [CrossRef]
- Murphy, S.M.C.; Bautista, M.A.; Cramm, M.A.; Hubert, C.R.J. Biodegradation of diesel and crude oil by Labrador Sea cold adapted microbial communities. Appl. Environ. Microbiol. 2021, 87, e0080021. [Google Scholar] [CrossRef]
- Brakstad, O.G.; Bonaunet, K. Biodegradation of petroleum hydrocarbons in seawater at low temperatures (0–5 °C) and bacterial communities associated with degradation. Biodegradation 2006, 17, 71–82. [Google Scholar] [CrossRef]
- Mapelli, F.; Scoma, A.; Michoud, G.; Aulenta, F.; Boon, N.; Borin, S.; Kalogerakis, N.; Daffonchio, D. Biotechnologies for marine oil spill cleanup: Indissoluble ties with microorganisms. Trends Biotechnol. 2017, 35, 860–870. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SW0 | SW4 | SW8 | SWO4 | SWO8 | SWOB4 | SWOB8 | |
|---|---|---|---|---|---|---|---|
| Colwellia | |||||||
| Kaiju | 3.96 | 4.94 | 3.86 | 4.57 | 10.87 | 1.73 | 0.60 |
| Kaiju/MAR | 17.78 | 8.46 | 5.18 | 4.90 | 13.63 | 1.86 | 1.08 |
| Kraken2 | 8.32 | 9.97 | 6.26 | 4.94 | 10.83 | 1.40 | 0.61 |
| Bracken | 6.73 | 8.16 | 5.34 | 4.33 | 10.05 | 1.33 | 0.54 |
| Amplicon | 6.45 | 4.63 | 3.53 | 2.75 | 9.91 | 1.07 | 1.80 |
| Quantification | NA | NA | 7.35 | NA | 13.83 | 0.85 | 0.34 |
| Cycloclasticus | |||||||
| Kaiju | 0.13 | 10.05 | 8.81 | 1.10 | 2.12 | 1.82 | 13.36 |
| Kaiju/MAR | 0.25 | 23.20 | 16.69 | 1.75 | 3.99 | 2.66 | 25.33 |
| Kraken2 | 0.18 | 1.90 | 1.48 | 0.14 | 0.23 | 0.21 | 4.88 |
| Bracken | 0.14 | 1.55 | 1.25 | 0.12 | 0.21 | 0.20 | 4.34 |
| Amplicon | 0.05 | 2.13 | 1.90 | 0.25 | 0.46 | 0.59 | 7.74 |
| Quantification | NA | NA | 0.25 | NA | 0.01 | 0.14 | 7.60 |
| Pseudomonas | |||||||
| Kaiju | 1.96 | 1.23 | 1.97 | 9.64 | 6.42 | 38.41 | 25.39 |
| Kaiju/MAR | 0.48 | 0.94 | 2.24 | 8.59 | 9.44 | 39.03 | 13.88 |
| Kraken2 | 3.02 | 5.68 | 5.37 | 13.64 | 13.06 | 50.54 | 34.33 |
| Bracken | 2.71 | 4.74 | 4.83 | 12.21 | 11.97 | 48.14 | 31.65 |
| Amplicon | 0.26 | 0.20 | 0.25 | 0.78 | 0.24 | 10.76 | 3.70 |
| Quantification | NA | NA | 2.59 | NA | 5.08 | 9.52 | 9.25 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nõlvak, H.; Dang, N.P.; Truu, M.; Peeb, A.; Tiirik, K.; O’Sadnick, M.; Truu, J. Microbial Community Dynamics during Biodegradation of Crude Oil and Its Response to Biostimulation in Svalbard Seawater at Low Temperature. Microorganisms 2021, 9, 2425. https://doi.org/10.3390/microorganisms9122425
Nõlvak H, Dang NP, Truu M, Peeb A, Tiirik K, O’Sadnick M, Truu J. Microbial Community Dynamics during Biodegradation of Crude Oil and Its Response to Biostimulation in Svalbard Seawater at Low Temperature. Microorganisms. 2021; 9(12):2425. https://doi.org/10.3390/microorganisms9122425
Chicago/Turabian StyleNõlvak, Hiie, Nga Phuong Dang, Marika Truu, Angela Peeb, Kertu Tiirik, Megan O’Sadnick, and Jaak Truu. 2021. "Microbial Community Dynamics during Biodegradation of Crude Oil and Its Response to Biostimulation in Svalbard Seawater at Low Temperature" Microorganisms 9, no. 12: 2425. https://doi.org/10.3390/microorganisms9122425