
A Mycobacteriophage-Based Vaccine Platform: SARS-CoV-2 Antigen Expression and Display

 , , , , and
, , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Bacterial Strains
2.2. Construction of Vaccine Candidates
2.3. Phage Amplification
2.4. Vaccine Stability Test
2.5. Mouse Studies
2.6. ELISA
2.7. Western Blot
2.8. Phage Neutralization Assay
2.9. Focus Reduction Neutralization Test (FRNT)
3. Results
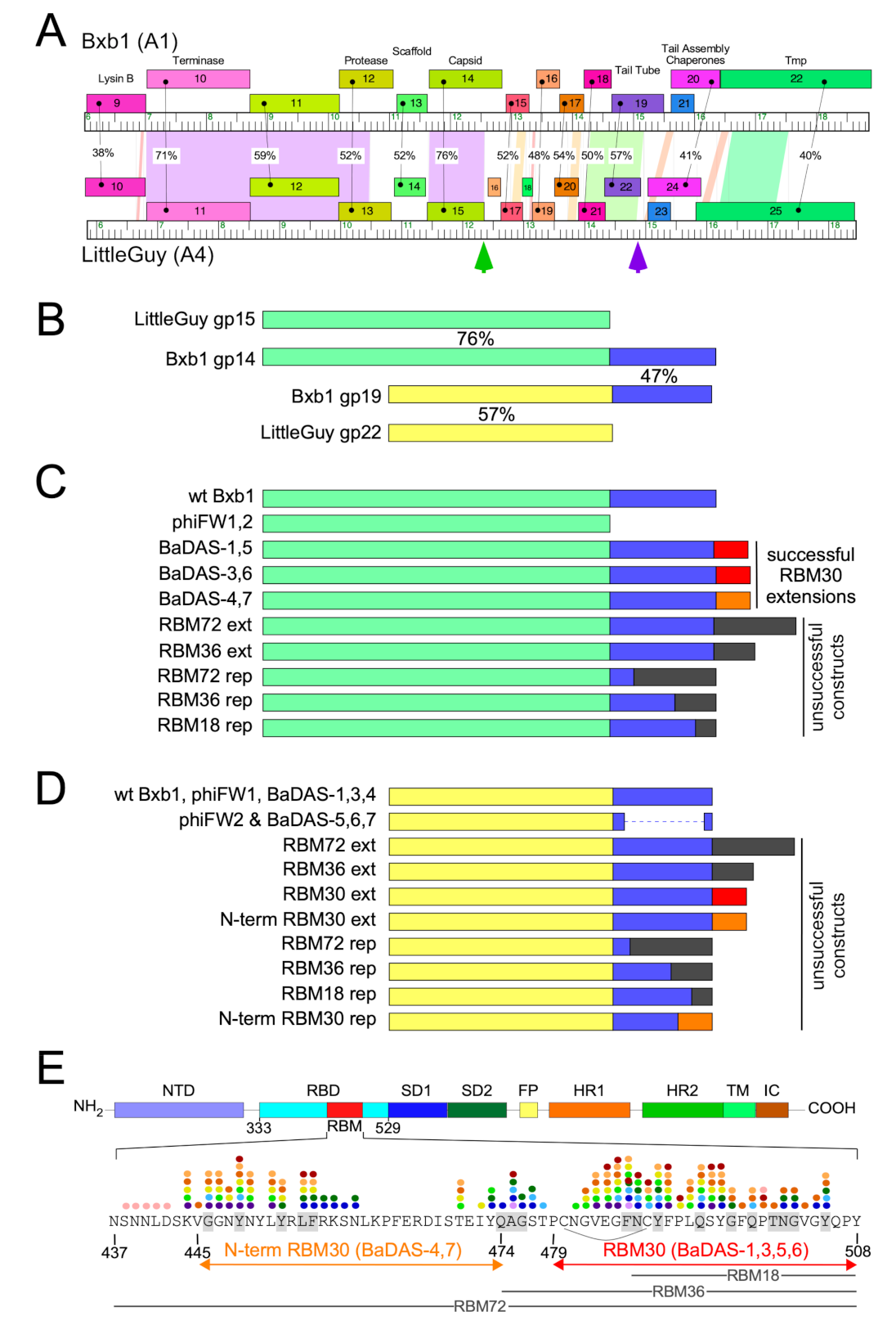
3.1. Mycobacteriophage phiTM45 as a Vaccine Platform Candidate
3.2. SARS-CoV-2 S Protein Epitopes for Display
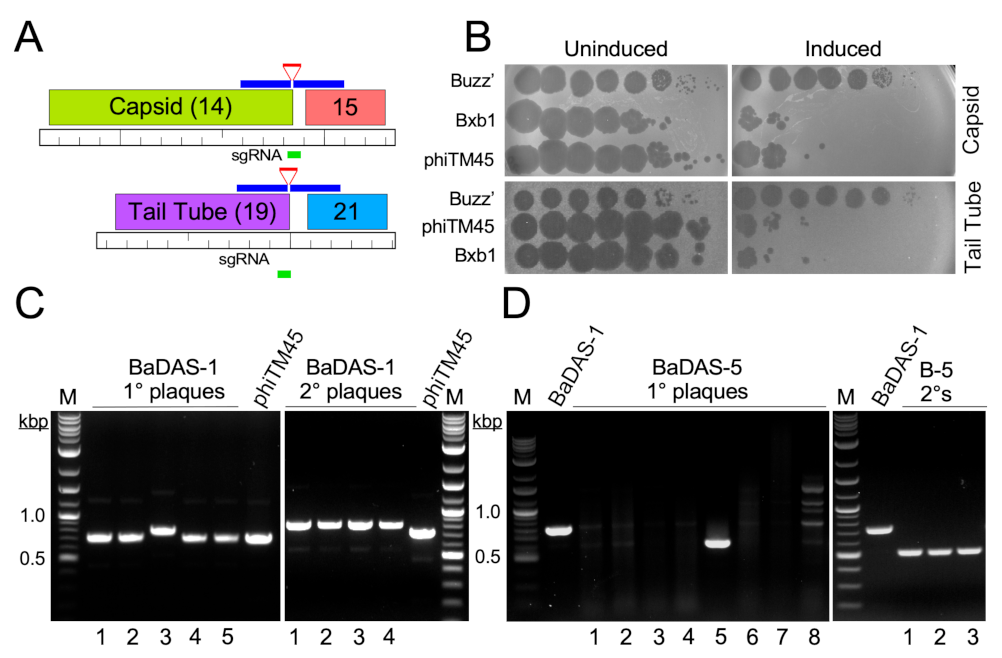
3.3. Construction of the BaDAS Series of Vaccine Candidates
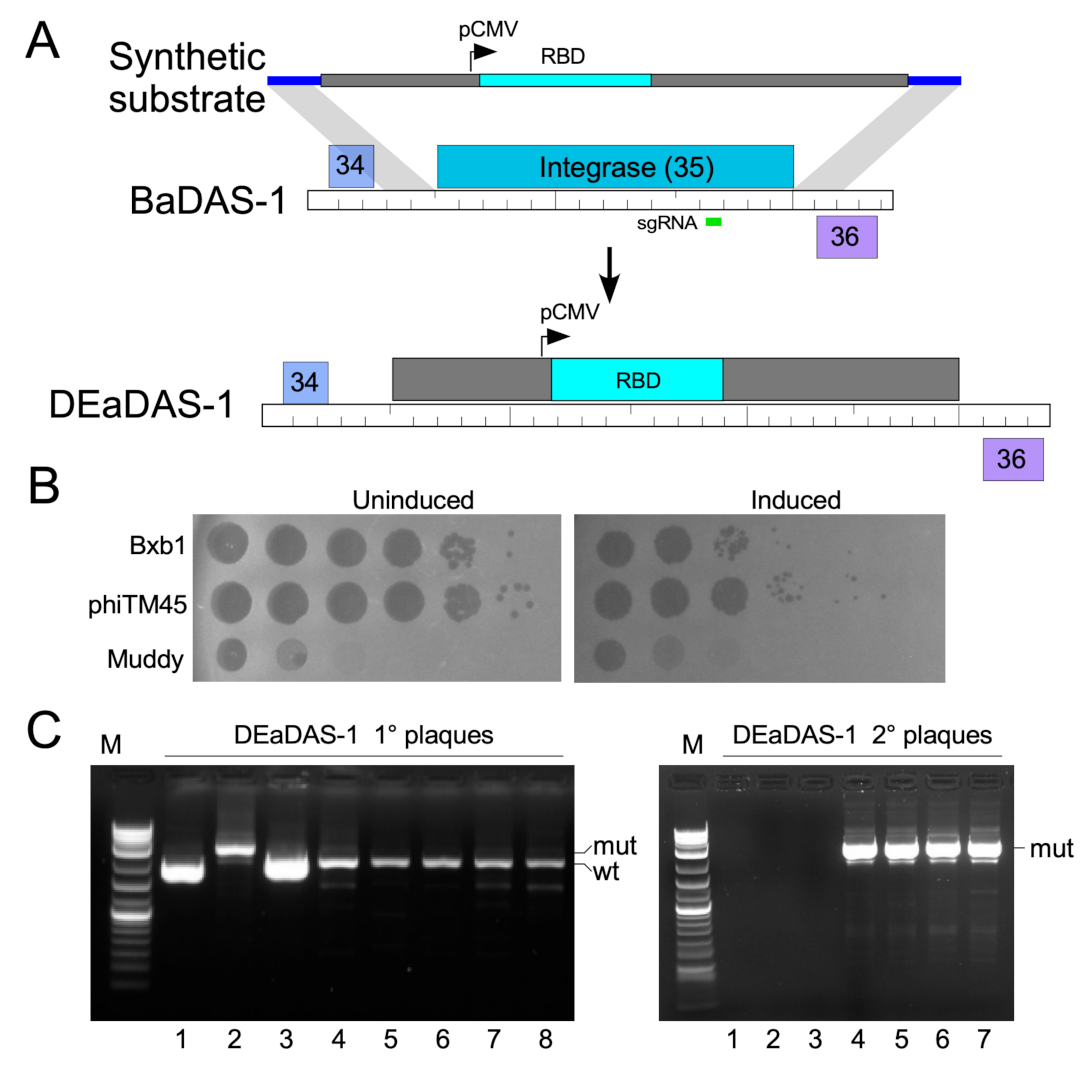
3.4. Construction of DEaDAS-1 Recombinant Vaccine Candidate
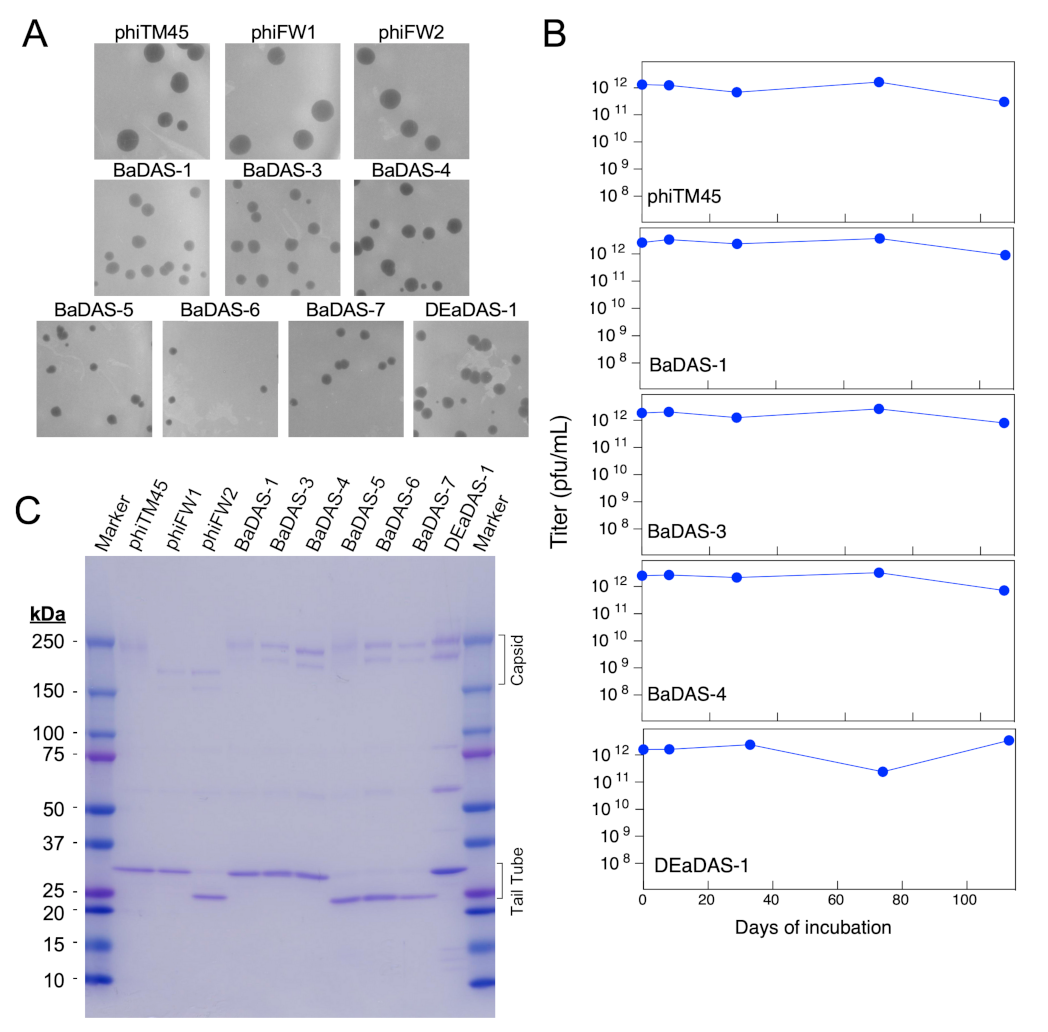
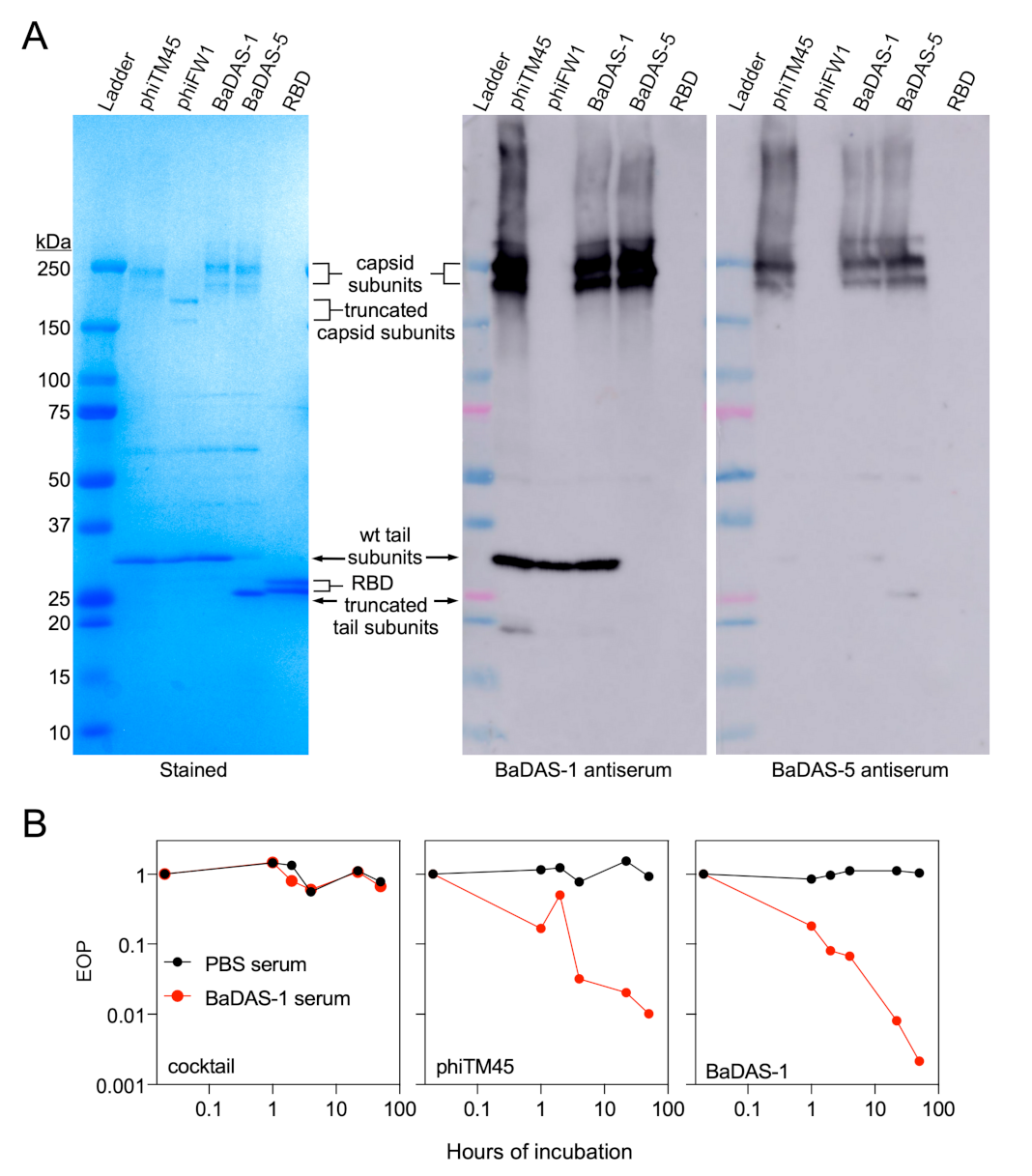
3.5. Characterization of Recombinant Phages
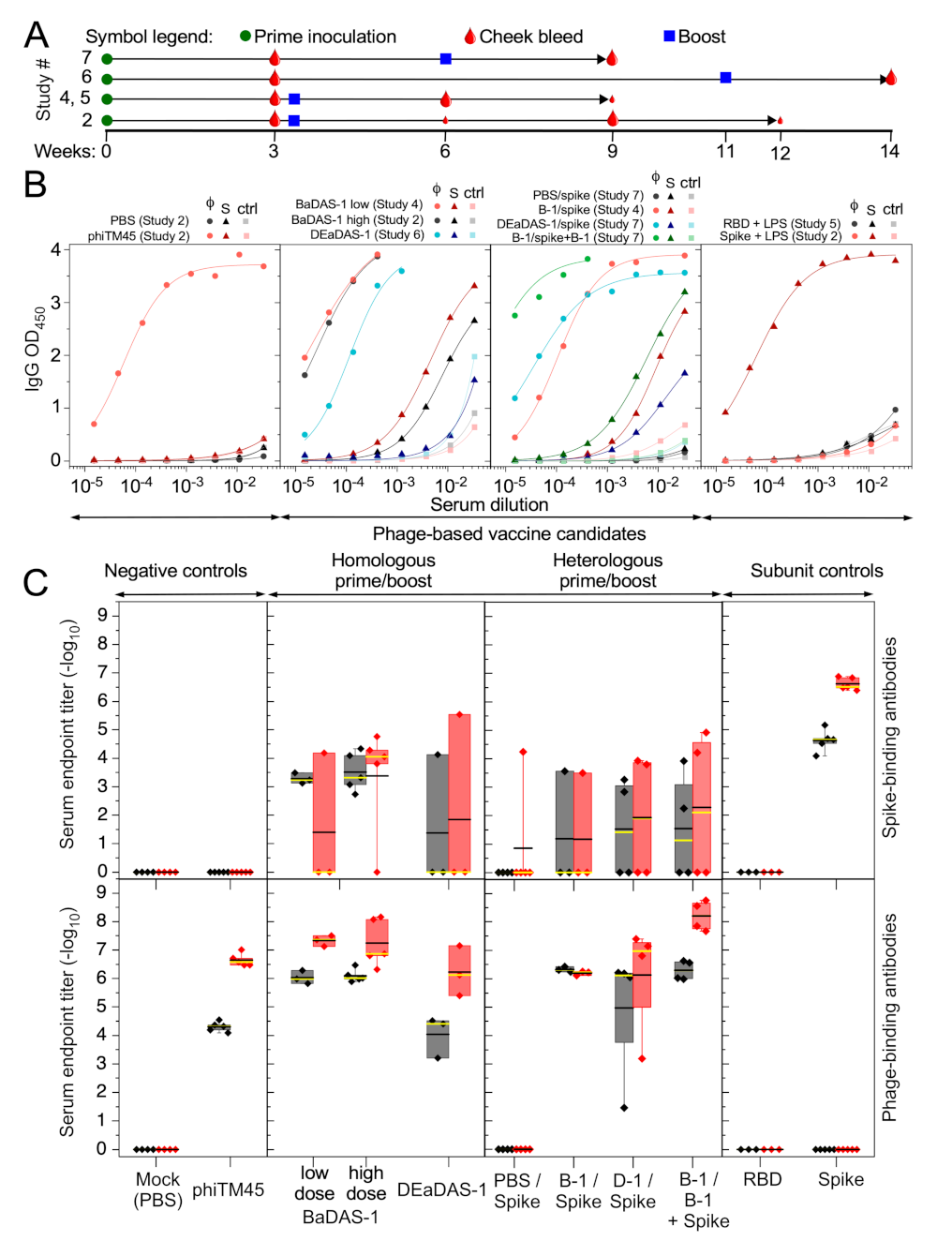
3.6. Immune Responses in Mice
3.7. Phage Capsid and Tail Tube Extensions Are Strongly Immunogenic
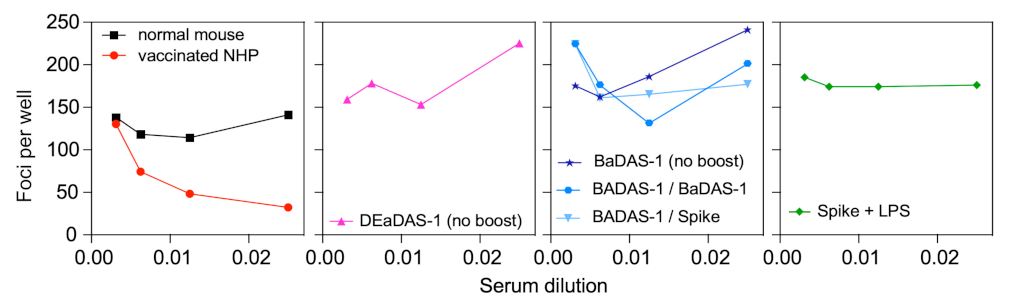
3.8. Spike-Binding Antibodies Raised in Mice Do Not Neutralize SARS-CoV-2 In Vitro
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gonzalez-Mora, A.; Hernandez-Perez, J.; Iqbal, H.M.N.; Rito-Palomares, M.; Benavides, J. Bacteriophage-Based Vaccines: A Potent Approach for Antigen Delivery. Vaccines 2020, 8, 504. [Google Scholar] [CrossRef]
- Haynes, B.F.; Corey, L.; Fernandes, P.; Gilbert, P.B.; Hotez, P.J.; Rao, S.; Santos, M.R.; Schuitemaker, H.; Watson, M.; Arvin, A. Prospects for a safe COVID-19 vaccine. Sci. Transl. Med. 2020, 12, eabe0948. [Google Scholar] [CrossRef] [PubMed]
- Saw, P.E.; Song, E.W. Phage display screening of therapeutic peptide for cancer targeting and therapy. Protein Cell 2019, 10, 787–807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, H.; Dong, S.; Zhang, X.; Chen, X.; Gao, X.; Wang, L. Phage vaccines displaying YGKDVKDLFDYAQE epitope induce protection against systemic candidiasis in mouse model. Vaccine 2018, 36, 5717–5724. [Google Scholar] [CrossRef]
- Bao, Q.; Li, X.; Han, G.; Zhu, Y.; Mao, C.; Yang, M. Phage-based vaccines. Adv. Drug Deliv. Rev. 2019, 145, 40–56. [Google Scholar] [CrossRef] [PubMed]
- Deng, L.; Ibanez, L.I.; Van den Bossche, V.; Roose, K.; Youssef, S.A.; de Bruin, A.; Fiers, W.; Saelens, X. Protection against Influenza A Virus Challenge with M2e-Displaying Filamentous Escherichia coli Phages. PLoS ONE 2015, 10, e0126650. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Tu, C.; Yu, X.; Zhang, M.; Zhang, N.; Zhao, M.; Nie, W.; Ren, Z. Bacteriophage T4 nanoparticle capsid surface SOC and HOC bipartite display with enhanced classical swine fever virus immunogenicity: A powerful immunological approach. J. Virol. Methods 2007, 139, 50–60. [Google Scholar] [CrossRef]
- Ren, Z.J.; Lewis, G.K.; Wingfield, P.T.; Locke, E.G.; Steven, A.C.; Black, L.W. Phage display of intact domains at high copy number: A system based on SOC, the small outer capsid protein of bacteriophage T4. Protein Sci. 1996, 5, 1833–1843. [Google Scholar] [CrossRef] [Green Version]
- Ren, Z.J.; Tian, C.J.; Zhu, Q.S.; Zhao, M.Y.; Xin, A.G.; Nie, W.X.; Ling, S.R.; Zhu, M.W.; Wu, J.Y.; Lan, H.Y.; et al. Orally delivered foot-and-mouth disease virus capsid protomer vaccine displayed on T4 bacteriophage surface: 100% protection from potency challenge in mice. Vaccine 2008, 26, 1471–1481. [Google Scholar] [CrossRef]
- Wong, C.L.; Sieo, C.C.; Tan, W.S. Display of the VP1 epitope of foot-and-mouth disease virus on bacteriophage T7 and its application in diagnosis. J. Virol. Methods 2013, 193, 611–619. [Google Scholar] [CrossRef]
- Wong, C.L.; Yong, C.Y.; Muhamad, A.; Syahir, A.; Omar, A.R.; Sieo, C.C.; Tan, W.S. A 12-residue epitope displayed on phage T7 reacts strongly with antibodies against foot-and-mouth disease virus. Appl. Microbiol. Biotechnol. 2018, 102, 4131–4142. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Cano, P.; Gamage, L.N.A.; Marciniuk, K.; Hayes, C.; Napper, S.; Hayes, S.; Griebel, P.J. Lambda display phage as a mucosal vaccine delivery vehicle for peptide antigens. Vaccine 2017, 35, 7256–7263. [Google Scholar] [CrossRef] [PubMed]
- Hayes, S. Bacterial Virus Lambda Gpd-Fusions to Cathelicidins, alpha- and beta-Defensins, and Disease-Specific Epitopes Evaluated for Antimicrobial Toxicity and Ability to Support Phage Display. Viruses 2019, 11, 869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayes, S.; Gamage, L.N.; Hayes, C. Dual expression system for assembling phage lambda display particle (LDP) vaccine to porcine Circovirus 2 (PCV2). Vaccine 2010, 28, 6789–6799. [Google Scholar] [CrossRef] [PubMed]
- Gamage, L.N.; Ellis, J.; Hayes, S. Immunogenicity of bacteriophage lambda particles displaying porcine Circovirus 2 (PCV2) capsid protein epitopes. Vaccine 2009, 27, 6595–6604. [Google Scholar] [CrossRef] [PubMed]
- Staquicini, D.I.; Tang, F.H.F.; Markosian, C.; Yao, V.J.; Staquicini, F.I.; Dodero-Rojas, E.; Contessoto, V.G.; Davis, D.; O'Brien, P.; Habib, N.; et al. Design and proof of concept for targeted phage-based COVID-19 vaccination strategies with a streamlined cold-free supply chain. Proc. Natl. Acad. Sci. USA 2021, 118, e2105739118. [Google Scholar] [CrossRef]
- Zhu, J.; Ananthaswamy, N.; Jain, S.; Batra, H.; Tang, W.C.; Lewry, D.A.; Richards, M.L.; David, S.A.; Kilgore, P.B.; Sha, J.; et al. A Universal Bacteriophage T4 Nanoparticle Platform to Design Multiplex SARS-CoV-2 Vaccine Candidates by CRISPR Engineering. bioRxiv 2021. [Google Scholar] [CrossRef]
- Dedrick, R.M.; Guerrero-Bustamante, C.A.; Garlena, R.A.; Russell, D.A.; Ford, K.; Harris, K.; Gilmour, K.C.; Soothill, J.; Jacobs-Sera, D.; Schooley, R.T.; et al. Engineered bacteriophages for treatment of a patient with a disseminated drug-resistant Mycobacterium abscessus. Nat. Med. 2019, 25, 730–733. [Google Scholar] [CrossRef]
- Schooley, R.T.; Biswas, B.; Gill, J.J.; Hernandez-Morales, A.; Lancaster, J.; Lessor, L.; Barr, J.J.; Reed, S.L.; Rohwer, F.; Benler, S.; et al. Development and Use of Personalized Bacteriophage-Based Therapeutic Cocktails To Treat a Patient with a Disseminated Resistant Acinetobacter baumannii Infection. Antimicrob. Agents Chemother. 2017, 61, e00954–17. [Google Scholar] [CrossRef] [Green Version]
- Schooley, R.T.; Strathdee, S. Treat phage like living antibiotics. Nat. Microbiol. 2020, 5, 391–392. [Google Scholar] [CrossRef]
- Russell, D.A.; Hatfull, G.F. PhagesDB: The actinobacteriophage database. Bioinformatics 2017, 33, 784–786. [Google Scholar] [CrossRef] [Green Version]
- Hatfull, G.F. Actinobacteriophages: Genomics, Dynamics, and Applications. Annu. Rev. Virol. 2020, 7, 37–61. [Google Scholar] [CrossRef]
- Lan, J.; Ge, J.; Yu, J.; Shan, S.; Zhou, H.; Fan, S.; Zhang, Q.; Shi, X.; Wang, Q.; Zhang, L.; et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 2020, 581, 215–220. [Google Scholar] [CrossRef] [Green Version]
- Corbett, K.S.; Edwards, D.K.; Leist, S.R.; Abiona, O.M.; Boyoglu-Barnum, S.; Gillespie, R.A.; Himansu, S.; Schafer, A.; Ziwawo, C.T.; DiPiazza, A.T.; et al. SARS-CoV-2 mRNA vaccine design enabled by prototype pathogen preparedness. Nature 2020, 586, 567–571. [Google Scholar] [CrossRef]
- Jackson, L.A.; Anderson, E.J.; Rouphael, N.G.; Roberts, P.C.; Makhene, M.; Coler, R.N.; McCullough, M.P.; Chappell, J.D.; Denison, M.R.; Stevens, L.J.; et al. An mRNA Vaccine against SARS-CoV-2 - Preliminary Report. N. Engl. J. Med. 2020, 383, 1920–1931. [Google Scholar] [CrossRef] [PubMed]
- Polack, F.P.; Thomas, S.J.; Kitchin, N.; Absalon, J.; Gurtman, A.; Lockhart, S.; Perez, J.L.; Perez Marc, G.; Moreira, E.D.; Zerbini, C.; et al. Safety and Efficacy of the BNT162b2 mRNA Covid-19 Vaccine. N. Engl. J. Med. 2020, 383, 2603–2615. [Google Scholar] [CrossRef] [PubMed]
- Folegatti, P.M.; Ewer, K.J.; Aley, P.K.; Angus, B.; Becker, S.; Belij-Rammerstorfer, S.; Bellamy, D.; Bibi, S.; Bittaye, M.; Clutterbuck, E.A.; et al. Safety and immunogenicity of the ChAdOx1 nCoV-19 vaccine against SARS-CoV-2: A preliminary report of a phase 1/2, single-blind, randomised controlled trial. Lancet 2020, 396, 467–478. [Google Scholar] [CrossRef]
- Tebas, P.; Yang, S.; Boyer, J.D.; Reuschel, E.L.; Patel, A.; Christensen-Quick, A.; Andrade, V.M.; Morrow, M.P.; Kraynyak, K.; Agnes, J.; et al. Safety and immunogenicity of INO-4800 DNA vaccine against SARS-CoV-2: A preliminary report of an open-label, Phase 1 clinical trial. EClinicalMedicine 2020, 31, 100689. [Google Scholar] [CrossRef] [PubMed]
- Guebre-Xabier, M.; Patel, N.; Tian, J.H.; Zhou, B.; Maciejewski, S.; Lam, K.; Portnoff, A.D.; Massare, M.J.; Frieman, M.B.; Piedra, P.A.; et al. NVX-CoV2373 vaccine protects cynomolgus macaque upper and lower airways against SARS-CoV-2 challenge. Vaccine 2020, 38, 7892–7896. [Google Scholar] [CrossRef]
- Liu, L.; Wang, P.; Nair, M.S.; Yu, J.; Rapp, M.; Wang, Q.; Luo, Y.; Chan, J.F.; Sahi, V.; Figueroa, A.; et al. Potent neutralizing antibodies against multiple epitopes on SARS-CoV-2 spike. Nature 2020, 584, 450–456. [Google Scholar] [CrossRef]
- Dedrick, R.M.; Freeman, K.G.; Nguyen, J.A.; Bahadirli-Talbott, A.; Smith, B.E.; Wu, A.E.; Ong, A.S.; Lin, C.T.; Ruppel, L.C.; Parrish, N.M.; et al. Potent antibody-mediated neutralization limits bacteriophage treatment of a pulmonary Mycobacterium abscessus infection. Nat. Med. 2021, 27, 1357–1361. [Google Scholar] [CrossRef] [PubMed]
- Broset, E.; Calvet Seral, J.; Arnal, C.; Uranga, S.; Kanno, A.I.; Leite, L.C.C.; Martin, C.; Gonzalo-Asensio, J. Engineering a new vaccine platform for heterologous antigen delivery in live-attenuated Mycobacterium tuberculosis. Comput. Struct. Biotechnol. J. 2021, 19, 4273–4283. [Google Scholar] [CrossRef]
- Stover, C.K.; de la Cruz, V.F.; Fuerst, T.R.; Burlein, J.E.; Benson, L.A.; Bennett, L.T.; Bansal, G.P.; Young, J.F.; Lee, M.H.; Hatfull, G.F.; et al. New use of BCG for recombinant vaccines. Nature 1991, 351, 456–460. [Google Scholar] [CrossRef] [PubMed]
- Mavrich, T.N.; Hatfull, G.F. Evolution of Superinfection Immunity in Cluster A Mycobacteriophages. mBio 2019, 10, e00971–19. [Google Scholar] [CrossRef] [Green Version]
- Jacobs-Sera, D.; Marinelli, L.J.; Bowman, C.; Broussard, G.W.; Guerrero Bustamante, C.; Boyle, M.M.; Petrova, Z.O.; Dedrick, R.M.; Pope, W.H.; Science Education Alliance Phage Hunters Advancing Genomics and Evolutionary Science program; et al. On the nature of mycobacteriophage diversity and host preference. Virology 2012, 434, 187–201. [Google Scholar] [CrossRef] [PubMed]
- Mediavilla, J.; Kriakov, J.; Ford, M.E.; Duda, R.L.; Jacobs, W.R.; Hendrix, R.W.; Hatfull, G.F. Genome organization and characterization of mycobacteriophage Bxb1. Mol. Microbiol. 2000, 38, 955–970. [Google Scholar] [CrossRef] [Green Version]
- Wetzel, K.S.; Guerrero-Bustamante, C.A.; Dedrick, R.M.; Ko, C.C.; Freeman, K.G.; Aull, H.G.; Divens, A.M.; Rock, J.M.; Zack, K.M.; Hatfull, G.F. Crispy-bred and crispy-brip: Efficient bacteriophage engineering. Sci. Rep. 2021, 11, 6796. [Google Scholar] [CrossRef]
- Marinelli, L.J.; Piuri, M.; Swigonova, Z.; Balachandran, A.; Oldfield, L.M.; van Kessel, J.C.; Hatfull, G.F. BRED: A simple and powerful tool for constructing mutant and recombinant bacteriophage genomes. PLoS ONE 2008, 3, e3957. [Google Scholar] [CrossRef] [Green Version]
- Russell, D.A. Sequencing, Assembling, and Finishing Complete Bacteriophage Genomes. Methods Mol. Biol. 2018, 1681, 109–125. [Google Scholar] [CrossRef]
- Xu, L.; Doyle, J.; Barbeau, D.J.; Le Sage, V.; Wells, A.; Duprex, W.P.; Shurin, M.R.; Wheeler, S.E.; McElroy, A.K. A Cross-Sectional Study of SARS-CoV-2 Seroprevalence between Fall 2020 and February 2021 in Allegheny County, Western Pennsylvania, USA. Pathogens 2021, 10, 710. [Google Scholar] [CrossRef]
- Hatfull, G.F. Mycobacteriophages. In The Bacteriophages; Calendar, R., Ed.; Oxford University Press: New York, NY, USA, 2006; pp. 602–620. [Google Scholar]
- Duda, R.L. Protein Chainmail: Catenated Protein in Viral Capsids. Cell 1998, 94, 55–60. [Google Scholar] [CrossRef] [Green Version]
- Wikoff, W.R.; Duda, R.L.; Hendrix, R.W.; Johnson, J.E. Crystallographic analysis of the dsDNA bacteriophage HK97 mature empty capsid. Acta Crystallogr. Sect. D Biol. Crystallogr. 1999, 55, 763–771. [Google Scholar] [CrossRef] [PubMed]
- Fraser, J.S.; Yu, Z.; Maxwell, K.L.; Davidson, A.R. Ig-like domains on bacteriophages: A tale of promiscuity and deceit. J. Mol. Biol. 2006, 359, 496–507. [Google Scholar] [CrossRef]
- Campbell, P.L.; Duda, R.L.; Nassur, J.; Conway, J.F.; Huet, A. Mobile Loops and Electrostatic Interactions Maintain the Flexible Tail Tube of Bacteriophage Lambda. J. Mol. Biol. 2020, 432, 384–395. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.M.; Wang, W.; Song, Z.G.; Hu, Y.; Tao, Z.W.; Tian, J.H.; Pei, Y.Y.; et al. A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef] [Green Version]
- Yan, R.; Zhang, Y.; Xia, L.; Guo, Y.; Zhou, Q. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science 2020, 367, 1444–1448. [Google Scholar] [CrossRef] [Green Version]
- Shang, J.; Ye, G.; Shi, K.; Wan, Y.; Luo, C.; Aihara, H.; Geng, Q.; Auerbach, A.; Li, F. Structural basis of receptor recognition by SARS-CoV-2. Nature 2020, 581, 221–224. [Google Scholar] [CrossRef] [Green Version]
- Ju, B.; Zhang, Q.; Ge, J.; Wang, R.; Sun, J.; Ge, X.; Yu, J.; Shan, S.; Zhou, B.; Song, S.; et al. Human neutralizing antibodies elicited by SARS-CoV-2 infection. Nature 2020, 584, 115–119. [Google Scholar] [CrossRef] [PubMed]
- Yi, C.; Sun, X.; Ye, J.; Ding, L.; Liu, M.; Yang, Z.; Lu, X.; Zhang, Y.; Ma, L.; Gu, W.; et al. Key residues of the receptor binding motif in the spike protein of SARS-CoV-2 that interact with ACE2 and neutralizing antibodies. Cell. Mol. Immunol. 2020, 17, 621–630. [Google Scholar] [CrossRef]
- Shi, R.; Shan, C.; Duan, X.; Chen, Z.; Liu, P.; Song, J.; Song, T.; Bi, X.; Han, C.; Wu, L.; et al. A human neutralizing antibody targets the receptor-binding site of SARS-CoV-2. Nature 2020, 584, 120–124. [Google Scholar] [CrossRef]
- Kreye, J.; Reincke, S.M.; Kornau, H.C.; Sanchez-Sendin, E.; Corman, V.M.; Liu, H.; Yuan, M.; Wu, N.C.; Zhu, X.; Lee, C.D.; et al. A Therapeutic Non-self-reactive SARS-CoV-2 Antibody Protects from Lung Pathology in a COVID-19 Hamster Model. Cell 2020, 183, 1058–1069. [Google Scholar] [CrossRef] [PubMed]
- Barnes, C.O.; Jette, C.A.; Abernathy, M.E.; Dam, K.A.; Esswein, S.R.; Gristick, H.B.; Malyutin, A.G.; Sharaf, N.G.; Huey-Tubman, K.E.; Lee, Y.E.; et al. SARS-CoV-2 neutralizing antibody structures inform therapeutic strategies. Nature 2020, 588, 682–687. [Google Scholar] [CrossRef] [PubMed]
- Letko, M.; Marzi, A.; Munster, V. Functional assessment of cell entry and receptor usage for SARS-CoV-2 and other lineage B betacoronaviruses. Nat. Microbiol. 2020, 5, 562–569. [Google Scholar] [CrossRef] [Green Version]
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [Green Version]
- Clark, J.R.; Bartley, K.; Jepson, C.D.; Craik, V.; March, J.B. Comparison of a bacteriophage-delivered DNA vaccine and a commercially available recombinant protein vaccine against hepatitis B. FEMS Immunol. Med. Microbiol. 2011, 61, 197–204. [Google Scholar] [CrossRef] [Green Version]
- Chan, K.K.; Dorosky, D.; Sharma, P.; Abbasi, S.A.; Dye, J.M.; Kranz, D.M.; Herbert, A.S.; Procko, E. Engineering human ACE2 to optimize binding to the spike protein of SARS coronavirus 2. Science 2020, 369, 6508. [Google Scholar] [CrossRef]
- Srivastava, V.; Niu, L.; Phadke, K.S.; Bellaire, B.H.; Cho, M.W. Induction of Potent and Durable Neutralizing Antibodies Against SARS-CoV-2 Using a Receptor Binding Domain-Based Immunogen. Front. Immunol. 2021, 12, 637982. [Google Scholar] [CrossRef]
- Lee, W.S.; Wheatley, A.K.; Kent, S.J.; DeKosky, B.J. Antibody-dependent enhancement and SARS-CoV-2 vaccines and therapies. Nat. Microbiol. 2020, 5, 1185–1191. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Edwards, R.J.; Manne, K.; Martinez, D.R.; Schäfer, A.; Alam, S.M.; Wiehe, K.; Lu, X.; Parks, R.; Sutherland, L.L.; et al. The functions of SARS-CoV-2 neutralizing and infection-enhancing antibodies in vitro and in mice and nonhuman primates. bioRxiv 2021. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Recombinant Phage | Description of Mutations |
|---|---|
| phiFW1 | gp14 truncated after D304 |
| phiFW2 | gp14 truncated after D304 gp19 truncated after V213 |
| BaDAS-1 | RBM30 inserted after gp14 S395 |
| BaDAS-3 | RBM30 inserted after gp14 G397 |
| BaDAS-4 | Nterm RBM30 inserted after gp14 G397 |
| BaDAS-5 | RBM30 inserted after gp14 S395 gp19 truncated after V213 |
| BaDAS-6 | RBM30 inserted after gp14 G397 gp19 truncated after V213 |
| BaDAS-7 | Nterm RBM30 inserted after gp14 G397 gp19 truncated after V213 |
| DEaDAS-1 | RBM30 inserted after gp14 S395 RBD expression cassette substituted for phiTM45 integrase, gene 35 |
| Study | Group | # of Mice/Group | Age of Mice (Weeks) | Sex of Mice | Prime | Prime Dose (pfu or µg) | Prime Adjuvant | Boost | Boost Dose(pfu or µg) | Boost Adjuvant | Bleed & Boost Schedule (Weeks Post Prime) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 1 | 4 | 15–20 | F M | PBS | N/A | N/A | PBS | N/A | N/A | 2—bleed 2—boost 4—bleed 4—boost 6—bleed 8—bleed |
| 2 | 4 | 15–20 | F M | phiTM45 | 1E10 | N/A | phiTM45 | 1E10 | N/A | ||
| 3 | 4 | 15–20 | F M | phiTM45 | 1E11 | N/A | phiTM45 | 1E11 | N/A | ||
| 4 | 4 | 15–20 | F M | BaDAS-1 | 1E10 | N/A | BaDAS-1 | 1E10 | N/A | ||
| 5 | 4 | 15–20 | F M | BaDAS-1 | 1E10 | 5 µg LPS | BaDAS-1 | 1E10 | 5 µg LPS | ||
| 6 | 4 | 15–20 | F M | BaDAS-1 | 1E11 | N/A | BaDAS-1 | 1E10 | N/A | ||
| 2 | 1 | 5 | 8 | F | PBS | N/A | N/A | PBS | N/A | N/A | 3—bleed 3—boost 6—bleed 9—bleed 12—bleed |
| 2 | 5 | 8 | F | Spike S1 | 20 | 5 µg LPS | Spike S1 | 20 | 5 µg LPS | ||
| 3 | 5 | 8 | F | phiTM45 | 1E11 | N/A | phiTM45 | 1E11 | N/A | ||
| 4 | 5 | 8 | F | phiTM45 | 1E11 | 5 µg LPS | phiTM45 | 1E11 | 5 µg LPS | ||
| 5 | 5 | 8 | F | BaDAS-1 | 1E11 | N/A | BaDAS-1 | 1E11 | N/A | ||
| 6 | 5 | 8 | F | BaDAS-1 | 1E11 | 5 µg LPS | BaDAS-1 | 1E11 | 5 µg LPS | ||
| 7 | 5 | 8 | F | BaDAS-3 | 1E11 | N/A | BaDAS-3 | 1E11 | N/A | ||
| 8 | 5 | 8 | F | BaDAS-3 | 1E11 | 5 µg LPS | BaDAS-3 | 1E11 | 5 µg LPS | ||
| 9 | 5 | 8 | F | BADAS-4 | 1E11 | N/A | BADAS-4 | 1E11 | N/A | ||
| 10 | 5 | 8 | F | BaDAS-4 | 1E11 | 5 µg LPS | BaDAS-4 | 1E11 | 5 µg LPS | ||
| 3 | 1 | 3 | 12 | F M | BADAS-5 | 5E10 | N/A | N/A | N/A | N/A | 2—bleed 4—bleed |
| 2 | 3 | 12 | F M | BaDAS-6 | 5E10 | N/A | N/A | N/A | N/A | ||
| 3 | 3 | 12 | F M | BaDAS-7 | 5E10 | N/A | N/A | N/A | N/A | ||
| 4 | 1 | 3 | 12 | F | BaDAS-1 | 5E10 | N/A | BaDAS-1 | 5E10 | N/A | 3—bleed 3—boost 6—bleed 9—bleed |
| 2 | 3 | 12 | F | BaDAS-1 | 5E10 | N/A | Spike S1 | 10 | N/A | ||
| 3 | 3 | 12 | F | BaDAS-5 | 5E10 | N/A | BaDAS-5 | 5E10 | N/A | ||
| 4 | 3 | 12 | F | BaDAS-5 | 5E10 | N/A | Spike S1 | 10 | N/A | ||
| 5 | 1 | 3 | 8 | F M | RBD | 2 | 2.5 µg LPS | RBD | 2 | 2.5 µg LPS | 3—bleed 3—boost 6—bleed 9—bleed |
| 6 | 1 | 3 | 12 | F | DEaDAS-1 | 5E9 | N/A | DEaDAS-1 | 1E11 | N/A | 3—bleed 6—bleed 11—boost 14—bleed |
| 7 | 1 | 4 | 8 | F | PBS | N/A | N/A | Spike S1 | 10 | N/A | 3—bleed 6—boost 9—bleed |
| 2 | 4 | 8 | F | BaDAS-1 | 1E11 | N/A | BaDAS-1 + Spike S1 | 1E11 + 10 | N/A | ||
| 3 | 4 | 8 | F | DEaDAS-1 | 1E11 | N/A | Spike S1 | 10 | N/A |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Freeman, K.G.; Wetzel, K.S.; Zhang, Y.; Zack, K.M.; Jacobs-Sera, D.; Walters, S.M.; Barbeau, D.J.; McElroy, A.K.; Williams, J.V.; Hatfull, G.F. A Mycobacteriophage-Based Vaccine Platform: SARS-CoV-2 Antigen Expression and Display. Microorganisms 2021, 9, 2414. https://doi.org/10.3390/microorganisms9122414
Freeman KG, Wetzel KS, Zhang Y, Zack KM, Jacobs-Sera D, Walters SM, Barbeau DJ, McElroy AK, Williams JV, Hatfull GF. A Mycobacteriophage-Based Vaccine Platform: SARS-CoV-2 Antigen Expression and Display. Microorganisms. 2021; 9(12):2414. https://doi.org/10.3390/microorganisms9122414
Chicago/Turabian StyleFreeman, Krista G., Katherine S. Wetzel, Yu Zhang, Kira M. Zack, Deborah Jacobs-Sera, Sara M. Walters, Dominique J. Barbeau, Anita K. McElroy, John V. Williams, and Graham F. Hatfull. 2021. "A Mycobacteriophage-Based Vaccine Platform: SARS-CoV-2 Antigen Expression and Display" Microorganisms 9, no. 12: 2414. https://doi.org/10.3390/microorganisms9122414