
Novel Cytoskeleton-Associated Proteins in Trypanosoma brucei Are Essential for Cell Morphogenesis and Cytokinesis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Trypanosome Cell Culture, Plasmid Construction and Transfection
2.2. HeLa Cell Culture and Transfection
2.3. Quantitative PCR
2.4. Fluorescence Microscopy
2.5. Protein Electrophoresis and Immunoblotting
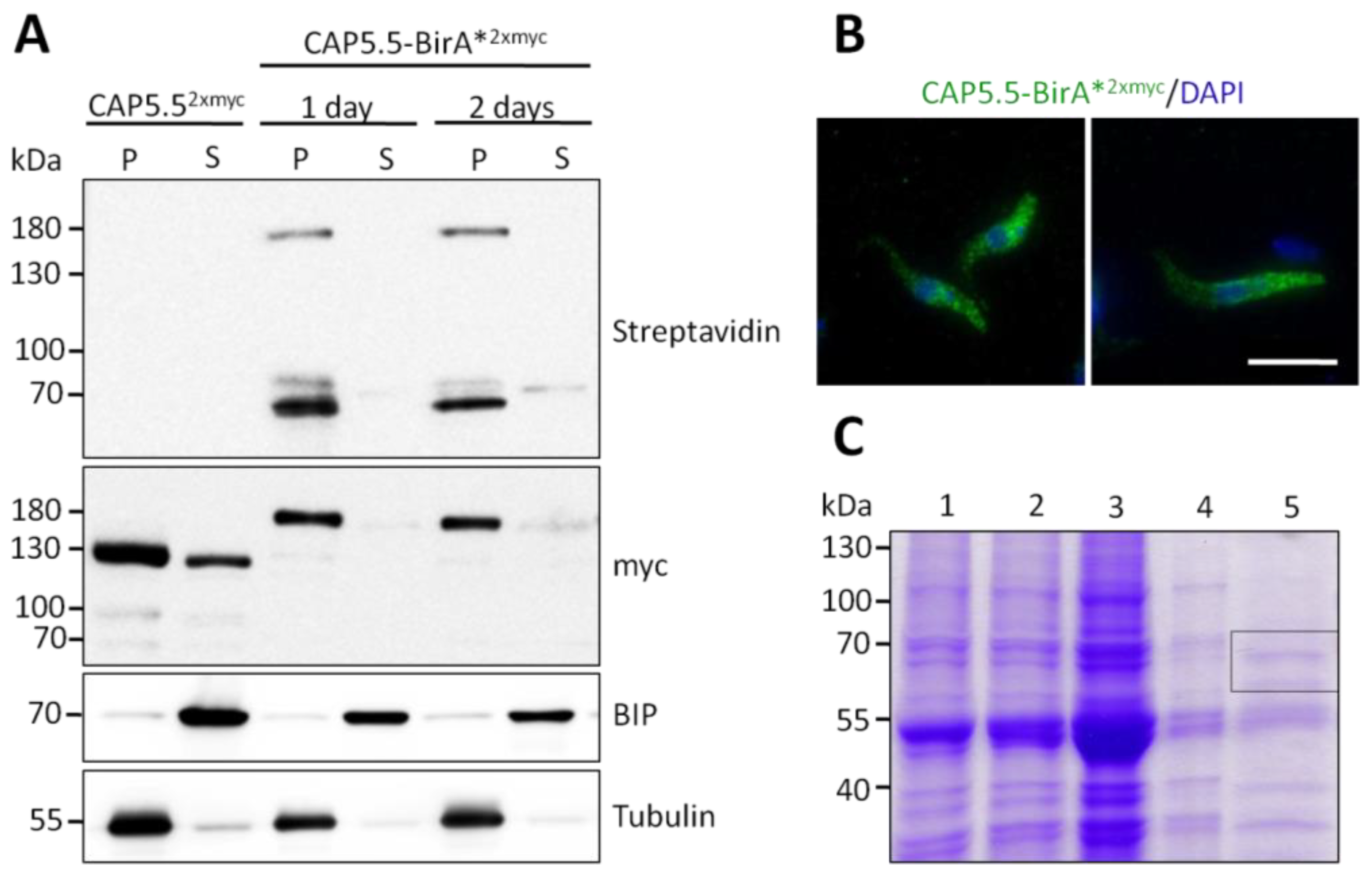
2.6. Purification of Biotinylated Proteins and Mass Spectrometry
2.7. Transmission Electron Microscopy
3. Results
3.1. Identification of Cytoskeleton-Associated Protein CAP50
3.2. CAP50 and CAP52 Are Related to Other T. brucei Cytoskeletal Proteins
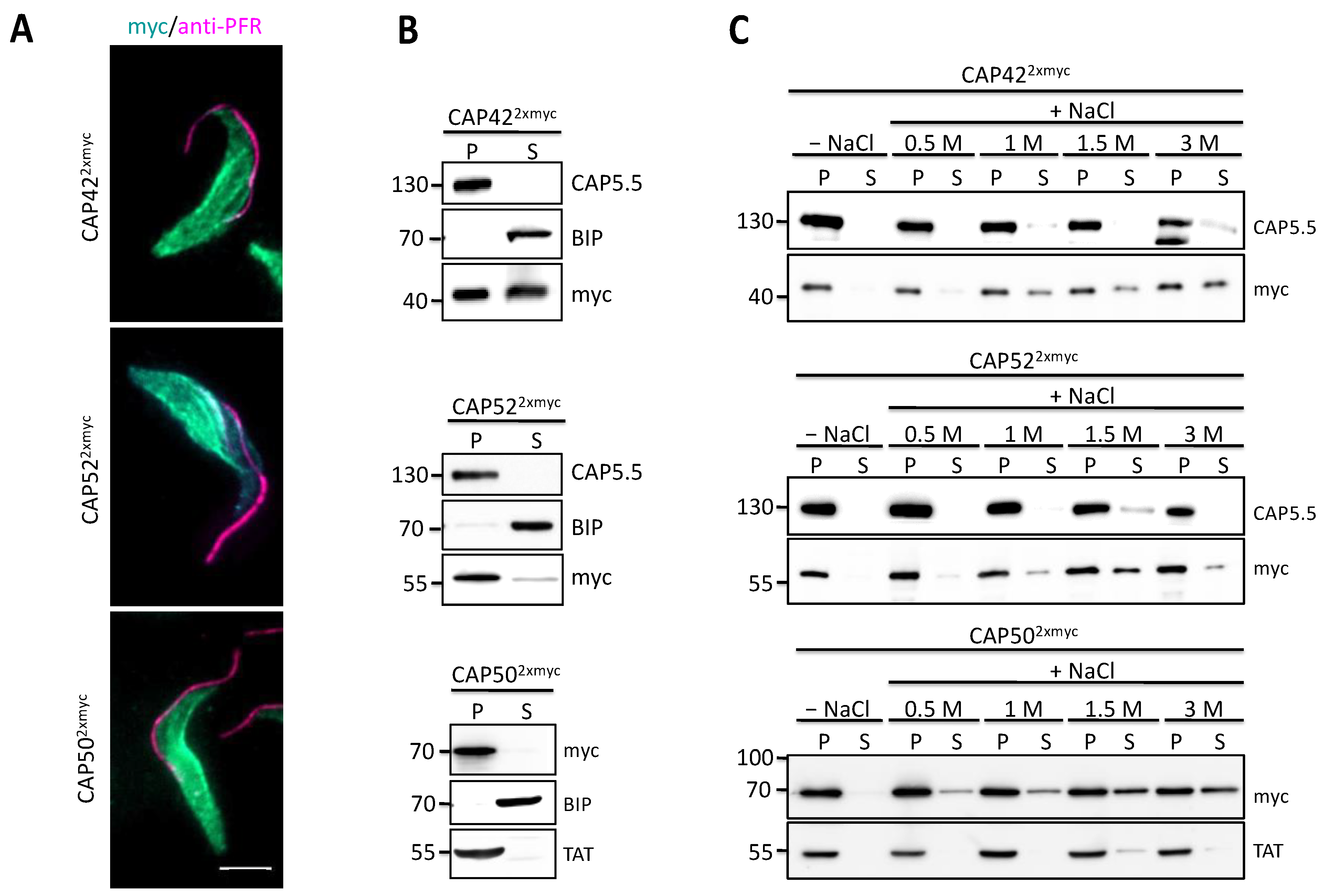
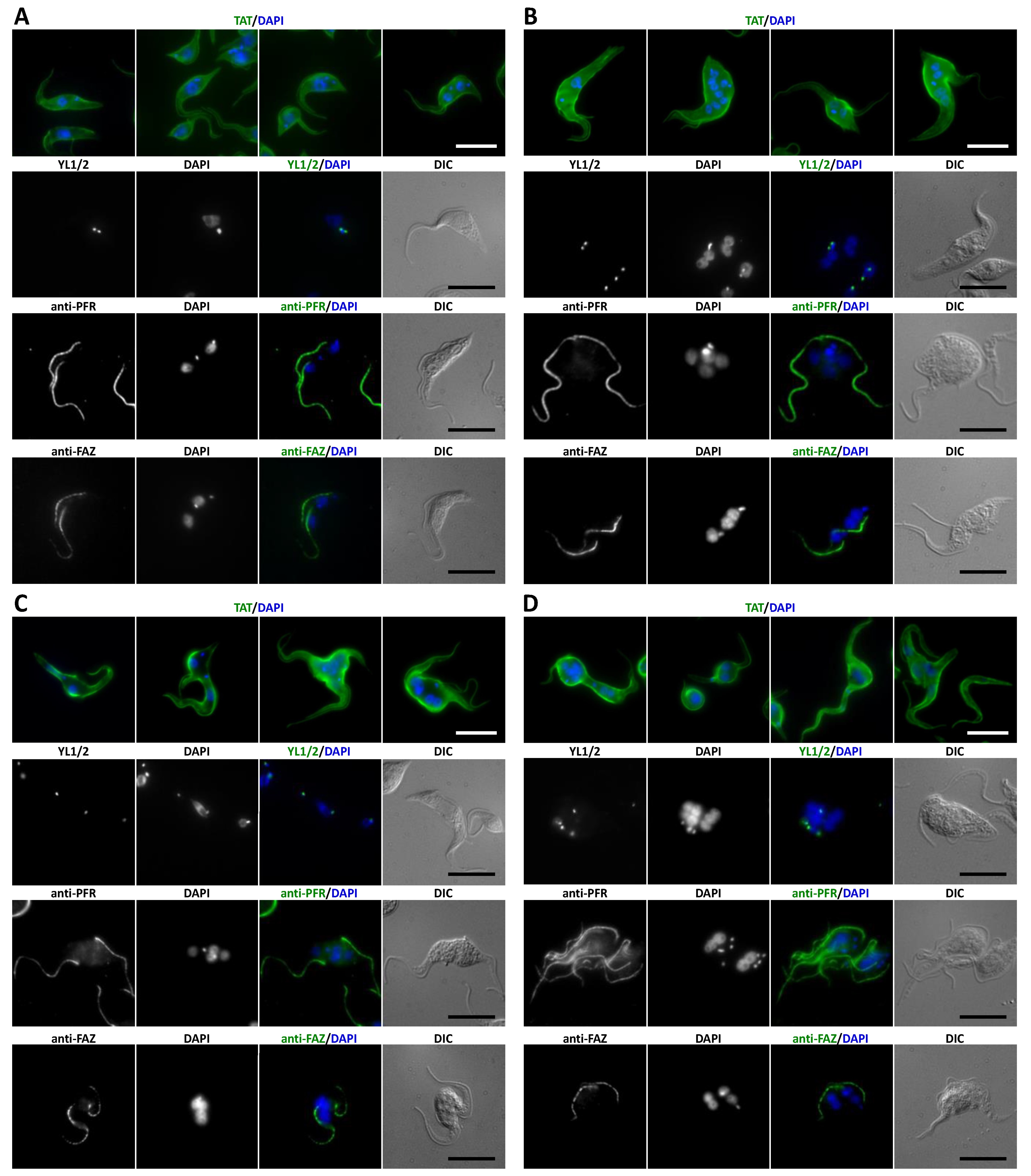
3.3. Novel CAPs Are Associated with the Subpellicular Microtubule Cytoskeleton
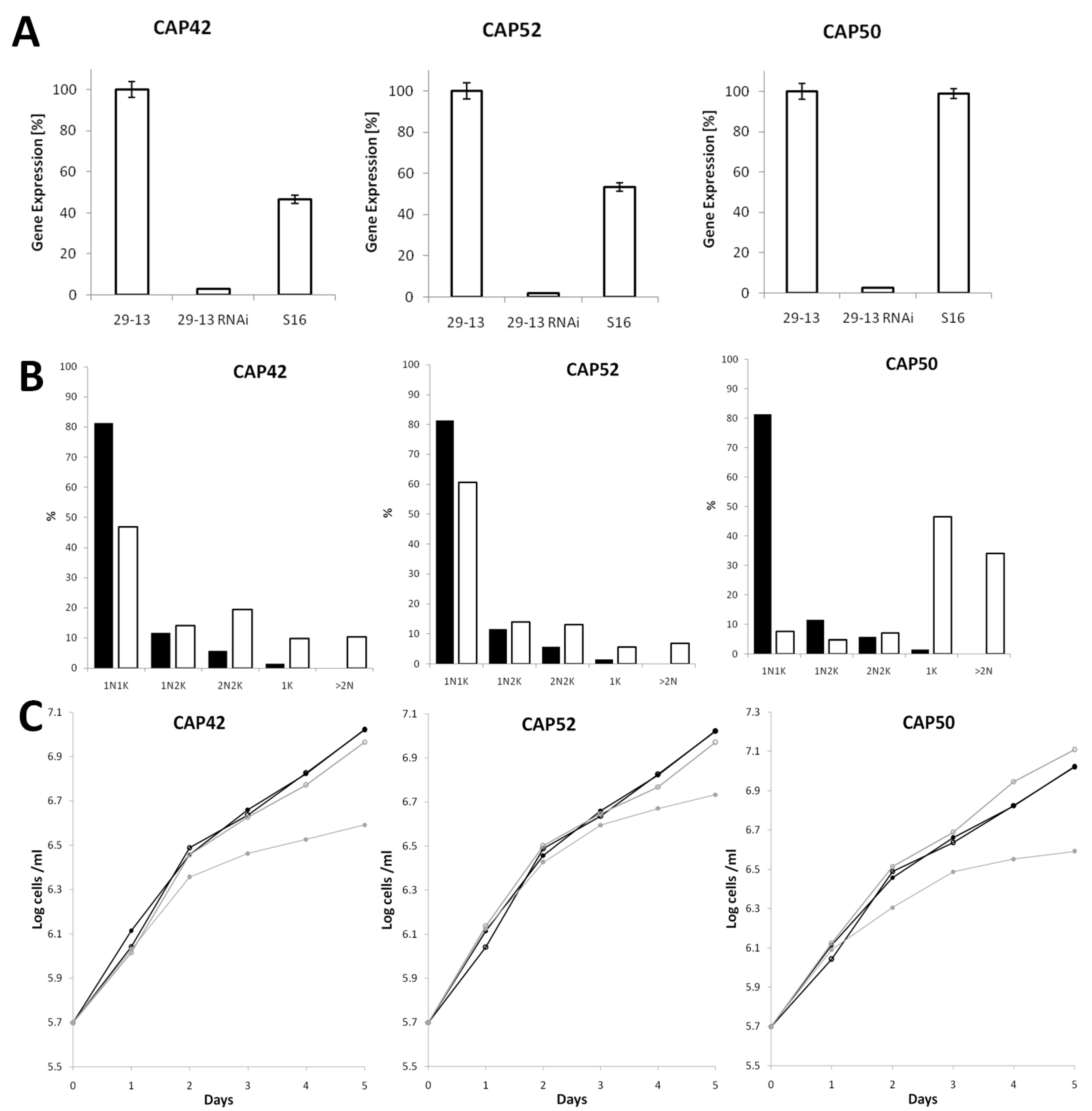
3.4. Depletion of CAP42, 50 and 52 Leads to Defects in Morphology, Cytokinesis and Growth
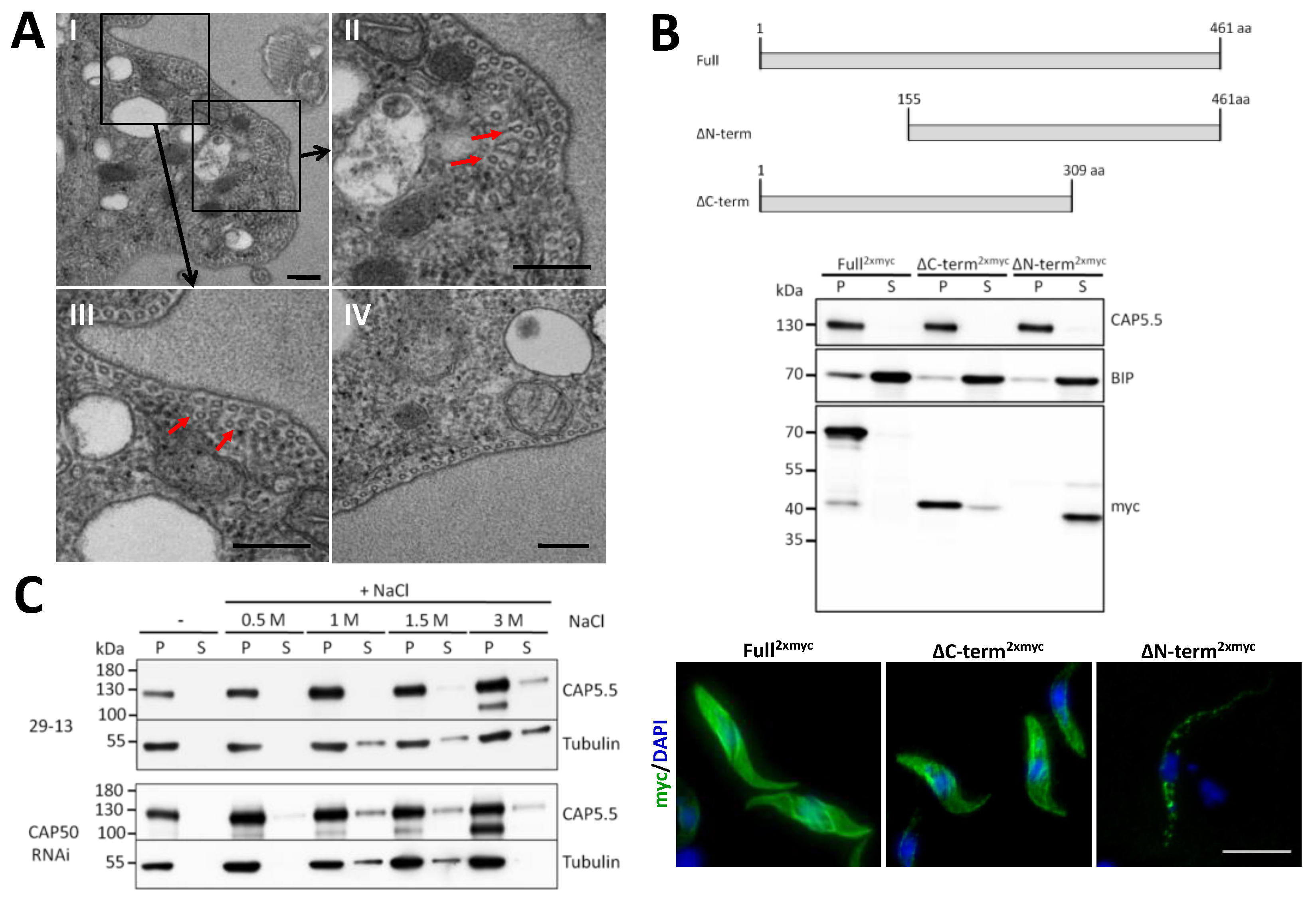
3.5. CAP50 Is Essential for Subpellicular Cytoskeleton Integrity
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jékely, G. Origin and evolution of the self-organizing cytoskeleton in the network of eukaryotic organelles. Cold Spring Harb. Perspect. Biol. 2014, 6, a016030. [Google Scholar] [CrossRef] [Green Version]
- Dawson, S.C.; Paredez, A.R. Alternative cytoskeletal landscapes: Cytoskeletal novelty and evolution in basal excavate protists. Curr. Opin. Cell Biol. 2013, 25, 134–141. [Google Scholar] [CrossRef] [Green Version]
- Gull, K. The cytoskeleton of trypanosomatid parasites. Annu. Rev. Microbiol. 1999, 53, 629–655. [Google Scholar] [CrossRef]
- Sinclair, A.N.; de Graffenried, C.L. More than microtubules: The structure and function of the subpellicular array in trypanosomatids. Trends Parasitol. 2019, 35, 760–777. [Google Scholar] [CrossRef] [PubMed]
- Hayes, P.; Varga, V.; Olego-Fernandez, S.; Sunter, J.; Ginger, M.L.; Gull, K. Modulation of a cytoskeletal calpain-like protein induces major transitions in trypanosome morphology. J. Cell Biol. 2014, 206, 377–384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wickstead, B.; Gull, K. The evolution of the cytoskeleton. J. Cell Biol. 2011, 194, 513–525. [Google Scholar] [CrossRef] [PubMed]
- Stevens, J.R.; Noyes, H.A.; Dover, G.A.; Gibson, W.C. The ancient and divergent origins of the human pathogenic trypanosomes, Trypanosoma brucei and T. cruzi. Parasitology 1999, 118, 107–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vickerman, K. Developmental cycles and biology of pathogenic trypanosomes. Br. Med. Bull. 1985, 41, 105–114. [Google Scholar] [CrossRef]
- Robinson, D.R.; Gull, K. Basal body movements as a mechanism for mitochondrial genome segregation in the trypanosome cell cycle. Nature 1991, 352, 731–733. [Google Scholar] [CrossRef]
- Bramblett, G.T.; Chang, S.L.; Flavin, M. Periodic crosslinking of microtubules by cytoplasmic microtubule-associated and microtubule-corset proteins from a trypanosomatid. Proc. Natl. Acad. Sci. USA 1987, 84, 3259–3263. [Google Scholar] [CrossRef] [Green Version]
- Hemphill, A.; Lawson, D.; Seebeck, T. The cytoskeletal architecture of Trypanosoma brucei. J. Parasitol. 1991, 77, 603–612. [Google Scholar] [CrossRef]
- Portman, N.; Gull, K. Identification of paralogous life-cycle stage specific cytoskeletal proteins in the parasite Trypanosoma brucei. PLoS ONE 2014, 9, e106777. [Google Scholar] [CrossRef] [PubMed]
- Sherwin, T.; Gull, K. Visualization of detyrosination along single microtubules reveals novel mechanisms of assembly during cytoskeletal duplication in trypanosomes. Cell 1989, 57, 211–221. [Google Scholar] [CrossRef]
- Hertz-Fowler, C.; Ersfeld, K.; Gull, K. CAP5.5, a life-cycle-regulated, cytoskeleton-associated protein is a member of a novel family of calpain-related proteins in Trypanosoma brucei. Mol. Biochem. Parasitol. 2001, 116, 25–34. [Google Scholar] [CrossRef]
- Liu, W.; Apagyi, K.; McLeavy, L.; Ersfeld, K. Expression and cellular localisation of calpain-like proteins in Trypanosoma brucei. Mol. Biochem. Parasitol. 2010, 169, 20–26. [Google Scholar] [CrossRef] [PubMed]
- Olego-Fernandez, S.; Vaughan, S.; Shaw, M.K.; Gull, K.; Ginger, M.L. Cell morphogenesis of Trypanosoma brucei requires the paralogous, differentially expressed calpain-related proteins CAP5.5 and CAP5.5V. Protist 2009, 160, 576–590. [Google Scholar] [CrossRef]
- Roux, K.J.; Kim, D.I.; Raida, M.; Burke, B. A promiscuous biotin ligase fusion protein identifies proximal and interacting proteins in mammalian cells. J. Cell Biol. 2012, 196, 801–810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roux, K.J.; Kim, D.I.; Burke, B.; May, D.G. BioID: A screen for protein-protein interactions. Curr. Protoc. Protein Sci. 2018, 91, 19–23. [Google Scholar] [CrossRef]
- Hu, H.; An, T.; Kurasawa, Y.; Zhou, Q.; Li, Z. The trypanosome-specific proteins FPRC and CIF4 regulate cytokinesis initiation by recruiting CIF1 to the cytokinesis initiation site. J. Biol. Chem. 2019, 294, 16672–16683. [Google Scholar] [CrossRef]
- Subota, I.; Julkowska, D.; Vincensini, L.; Reeg, N.; Buisson, J.; Blisnick, T.; Huet, D.; Perrot, S.; Santi-Rocca, J.; Duchateau, M.; et al. Proteomic analysis of intact flagella of procyclic Trypanosoma brucei cells identifies novel flagellar proteins with unique sub-localization and dynamics. Mol. Cell. Proteom. 2014, 13, 1769–1786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Q.; Hu, H.; He, C.Y.; Li, Z. Assembly and maintenance of the flagellum attachment zone filament in Trypanosoma brucei. J. Cell Sci. 2015, 128, 2361–2372. [Google Scholar] [CrossRef] [Green Version]
- Wirtz, E.; Clayton, C. Inducible gene expression in trypanosomes mediated by a prokaryotic repressor. Science 1995, 268, 1179–1182. [Google Scholar] [CrossRef] [PubMed]
- Wirtz, E.; Leal, S.; Ochatt, C.; Cross, G.A. A tightly regulated inducible expression system for conditional gene knock-outs and dominant-negative genetics in Trypanosoma brucei. Mol. Biochem. Parasitol. 1999, 99, 89–101. [Google Scholar] [CrossRef]
- Brun, R.; Schönenburger, M. Cultivation and in vitro cloning of procyclyc forms of Trypanosoma brucei in a semi-defined medium. Acta Trop. 1979, 36, 289–292. [Google Scholar]
- Morriswood, B.; Havlicek, K.; Demmel, L.; Yavuz, S.; Sealey-Cardona, M.; Vidilaseris, K.; Anrather, D.; Kostan, J.; Djinovic-Carugo, K.; Roux, K.J.; et al. Novel bilobe components in Trypanosoma brucei identified using proximity-dependent biotinylation. Eukaryot. Cell 2013, 12, 356–367. [Google Scholar] [CrossRef] [Green Version]
- Colasante, C.; Alibu, V.P.; Kirchberger, S.; Tjaden, J.; Clayton, C.; Voncken, F. Characterization and developmentally regulated localization of the mitochondrial carrier protein homologue MCP6 from Trypanosoma brucei. Eukaryot. Cell 2006, 5, 1194–1205. [Google Scholar] [CrossRef] [Green Version]
- Bochud-Allemann, N.; Schneider, A. Mitochondrial substrate level phosphorylation is essential for growth of procyclic Trypanosoma brucei. J. Biol. Chem. 2002, 277, 32849–32854. [Google Scholar] [CrossRef] [Green Version]
- Burkard, G.; Fragoso, C.M.; Roditi, I. Highly efficient stable transformation of bloodstream forms of Trypanosoma brucei. Mol. Biochem. Parasitol. 2007, 153, 220–223. [Google Scholar] [CrossRef] [PubMed]
- Schumann Burkard, G.; Jutzi, P.; Roditi, I. Genome-wide RNAi screens in bloodstream form trypanosomes identify drug transporters. Mol. Biochem. Parasitol. 2011, 175, 91–94. [Google Scholar] [CrossRef] [PubMed]
- Kohl, L.; Sherwin, T.; Gull, K. Assembly of the paraflagellar rod and the flagellum attachment zone complex during the Trypanosoma brucei cell cycle. J. Eukaryot. Microbiol. 1999, 46, 105–109. [Google Scholar] [CrossRef]
- Kilmartin, J.V.; Wright, B.; Milstein, C. Rat monoclonal antitubulin antibodies derived by using a new nonsecreting rat cell line. J. Cell Biol. 1982, 93, 576–582. [Google Scholar] [CrossRef]
- Woods, A.; Sherwin, T.; Sasse, R.; MacRae, T.H.; Baines, A.J.; Gull, K. Definition of individual components within the cytoskeleton of Trypanosoma brucei by a library of monoclonal antibodies. J. Cell Sci. 1989, 93, 491–500. [Google Scholar] [CrossRef] [PubMed]
- Wessel, D.; Flugge, U.I. A method for the quantitative recovery of protein in dilute solution in the presence of detergents and lipids. Anal. Biochem. 1984, 138, 141–143. [Google Scholar] [CrossRef]
- Winter, I.; Lockhauserbaumer, J.; Lallinger-Kube, G.; Schobert, R.; Ersfeld, K.; Biersack, B. Anti-trypanosomal activity of cationic N-heterocyclic carbene gold(I) complexes. Mol. Biochem. Parasitol. 2017, 214, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Shevchenko, A.; Wilm, M.; Vorm, O.; Mann, M. Mass spectrometric sequencing of proteins silver-stained polyacrylamide gels. Anal. Chem. 1996, 68, 850–858. [Google Scholar] [CrossRef]
- Hilton, N.A.; Sladewski, T.E.; Perry, J.A.; Pataki, Z.; Sinclair-Davis, A.N.; Muniz, R.S.; Tran, H.L.; Wurster, J.I.; Seo, J.; de Graffenried, C.L. Identification of TOEFAZ1-interacting proteins reveals key regulators of Trypanosoma brucei cytokinesis. Mol. Microbiol. 2018, 109, 306–326. [Google Scholar] [CrossRef] [Green Version]
- Buschmann, H.; Chan, J.; Sanchez-Pulido, L.; Andrade-Navarro, M.A.; Doonan, J.H.; Lloyd, C.W. Microtubule-associated AIR9 recognizes the cortical division site at preprophase and cell-plate insertion. Curr. Biol. 2006, 16, 1938–1943. [Google Scholar] [CrossRef] [Green Version]
- Buschmann, H.; Sanchez-Pulido, L.; Andrade-Navarro, M.A.; Lloyd, C.W. Homologues of Arabidopsis Microtubule-Associated AIR9 in Trypanosomatid Parasites: Hints on Evolution and Function. Plant Signal. Behav. 2007, 2, 296–299. [Google Scholar] [CrossRef] [Green Version]
- May, S.F.; Peacock, L.; Almeida Costa, C.I.; Gibson, W.C.; Tetley, L.; Robinson, D.R.; Hammarton, T.C. The Trypanosoma brucei AIR9-like protein is cytoskeleton-associated and is required for nucleus positioning and accurate cleavage furrow placement. Mol. Microbiol. 2012, 84, 77–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dean, S.; Sunter, J.D.; Wheeler, R.J. TrypTag.org: A Trypanosome Genome-wide Protein Localisation Resource. Trends Parasitol. 2017, 33, 80–82. [Google Scholar] [CrossRef] [Green Version]
- Humm, A.; Fritsche, E.; Steinbacher, S. Structure and reaction mechanism of L-arginine:glycine amidinotransferase. Biol. Chem. 1997, 378, 193–197. [Google Scholar]
- Fritsche, E.; Bergner, A.; Humm, A.; Piepersberg, W.; Huber, R. Crystal structure of L-arginine:inosamine-phosphate amidinotransferase StrB1 from Streptomyces griseus: An enzyme involved in streptomycin biosynthesis. Biochemistry 1998, 37, 17664–17672. [Google Scholar] [CrossRef]
- McAllaster, M.R.; Ikeda, K.N.; Lozano-Nunez, A.; Anrather, D.; Unterwurzacher, V.; Gossenreiter, T.; Perry, J.A.; Crickley, R.; Mercadante, C.J.; Vaughan, S.; et al. Proteomic identification of novel cytoskeletal proteins associated with TbPLK, an essential regulator of cell morphogenesis in Trypanosoma brucei. Mol. Biol. Cell 2015, 26, 3013–3029. [Google Scholar] [CrossRef]
- Zhou, Q.; Gu, J.; Lun, Z.R.; Ayala, F.J.; Li, Z. Two distinct cytokinesis pathways drive trypanosome cell division initiation from opposite cell ends. Proc. Natl. Acad. Sci. USA 2016, 113, 3287–3292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lacomble, S.; Vaughan, S.; Gadelha, C.; Morphew, M.K.; Shaw, M.K.; McIntosh, J.R.; Gull, K. Three-dimensional cellular architecture of the flagellar pocket and associated cytoskeleton in trypanosomes revealed by electron microscope tomography. J. Cell Sci. 2009, 122, 1081–1090. [Google Scholar] [CrossRef] [Green Version]
- Sunter, J.D.; Gull, K. The flagellum attachment zone: ‘The cellular ruler’ of trypanosome morphology. Trends Parasitol. 2016, 32, 309–324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uversky, V.N. What does it mean to be natively unfolded? Eur. J. Biochem. 2002, 269, 2–12. [Google Scholar] [CrossRef]
- Affolter, M.; Hemphill, A.; Roditi, I.; Muller, N.; Seebeck, T. The repetitive microtubule-associated proteins MARP-1 and MARP-2 of Trypanosoma brucei. J. Struct. Biol. 1994, 112, 241–251. [Google Scholar] [CrossRef]
- Baines, A.; Gull, K. WCB is a C2 domain protein defining the plasma membrane-sub-pellicular microtubule corset of kinetoplastid parasites. Protist 2008, 159, 115–125. [Google Scholar] [CrossRef]
- Detmer, E.; Hemphill, A.; Muller, N.; Seebeck, T. The Trypanosoma brucei autoantigen 1/6 is an internally repetitive cytoskeletal protein. Eur. J. Cell Biol. 1997, 72, 378–384. [Google Scholar] [PubMed]
- Rindisbacher, L.; Hemphill, A.; Seebeck, T. A repetitive protein from Trypanosome brucei which caps the microtubules at the posterior end of the cytoskeleton. Mol. Biochem. Parasitol. 1993, 58, 83–96. [Google Scholar] [CrossRef]
- Hemphill, A.; Affolter, M.; Seebeck, T. A novel microtubule-binding motif identified in a high molecular weight microtubule-associated protein from Trypanosoma brucei. J. Cell Biol. 1992, 117, 95–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alsford, S.; Turner, D.J.; Obado, S.O.; Sanchez-Flores, A.; Glover, L.; Berriman, M.; Hertz-Fowler, C.; Horn, D. High-throughput phenotyping using parallel sequencing of RNA interference targets in the African trypanosome. Genome Res. 2011, 21, 915–924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schimke, R.T.; Berlin, C.M.; Sweeney, E.W.; Carroll, W.R. The generation of energy by the arginine dihydrolase pathway in Mycoplasma hominis 07. J. Biol. Chem. 1966, 241, 2228–2236. [Google Scholar] [CrossRef]
- Tilvawala, R.; Thompson, P.R. Peptidyl arginine deiminases: Detection and functional analysis of protein citrullination. Curr. Opin. Struct. Biol. 2019, 59, 205–215. [Google Scholar] [CrossRef] [PubMed]
- Yabu, Y.; Koide, T.; Ohta, N.; Nose, M.; Ogihara, Y. Continuous growth of bloodstream forms of Trypanosoma brucei brucei in an axenic culture system containing a low concentration of serum. Southeast Asian J. Trop. Med. Public Health 1998, 29, 591–595. [Google Scholar] [PubMed]
- Vedrenne, C.; Giroud, C.; Robinson, D.R.; Besteiro, S.; Bosc, C.; Bringaud, F.; Baltz, T. Two related subpellicular cytoskeleton-associated proteins in Trypanosoma brucei stabilize microtubules. Mol. Biol. Cell 2002, 13, 1058–1070. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schock, M.; Schmidt, S.; Ersfeld, K. Novel Cytoskeleton-Associated Proteins in Trypanosoma brucei Are Essential for Cell Morphogenesis and Cytokinesis. Microorganisms 2021, 9, 2234. https://doi.org/10.3390/microorganisms9112234
Schock M, Schmidt S, Ersfeld K. Novel Cytoskeleton-Associated Proteins in Trypanosoma brucei Are Essential for Cell Morphogenesis and Cytokinesis. Microorganisms. 2021; 9(11):2234. https://doi.org/10.3390/microorganisms9112234
Chicago/Turabian StyleSchock, Marina, Steffen Schmidt, and Klaus Ersfeld. 2021. "Novel Cytoskeleton-Associated Proteins in Trypanosoma brucei Are Essential for Cell Morphogenesis and Cytokinesis" Microorganisms 9, no. 11: 2234. https://doi.org/10.3390/microorganisms9112234