Whole Genome Sequencing Characterization of HEV3-e and HEV3-f Subtypes among the Wild Boar Population in the Abruzzo Region, Italy: First Report

 , , , , ,
, , , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Processing and RNA Extraction
2.2. Real-Time RT–PCR Assay
2.3. Sequencing
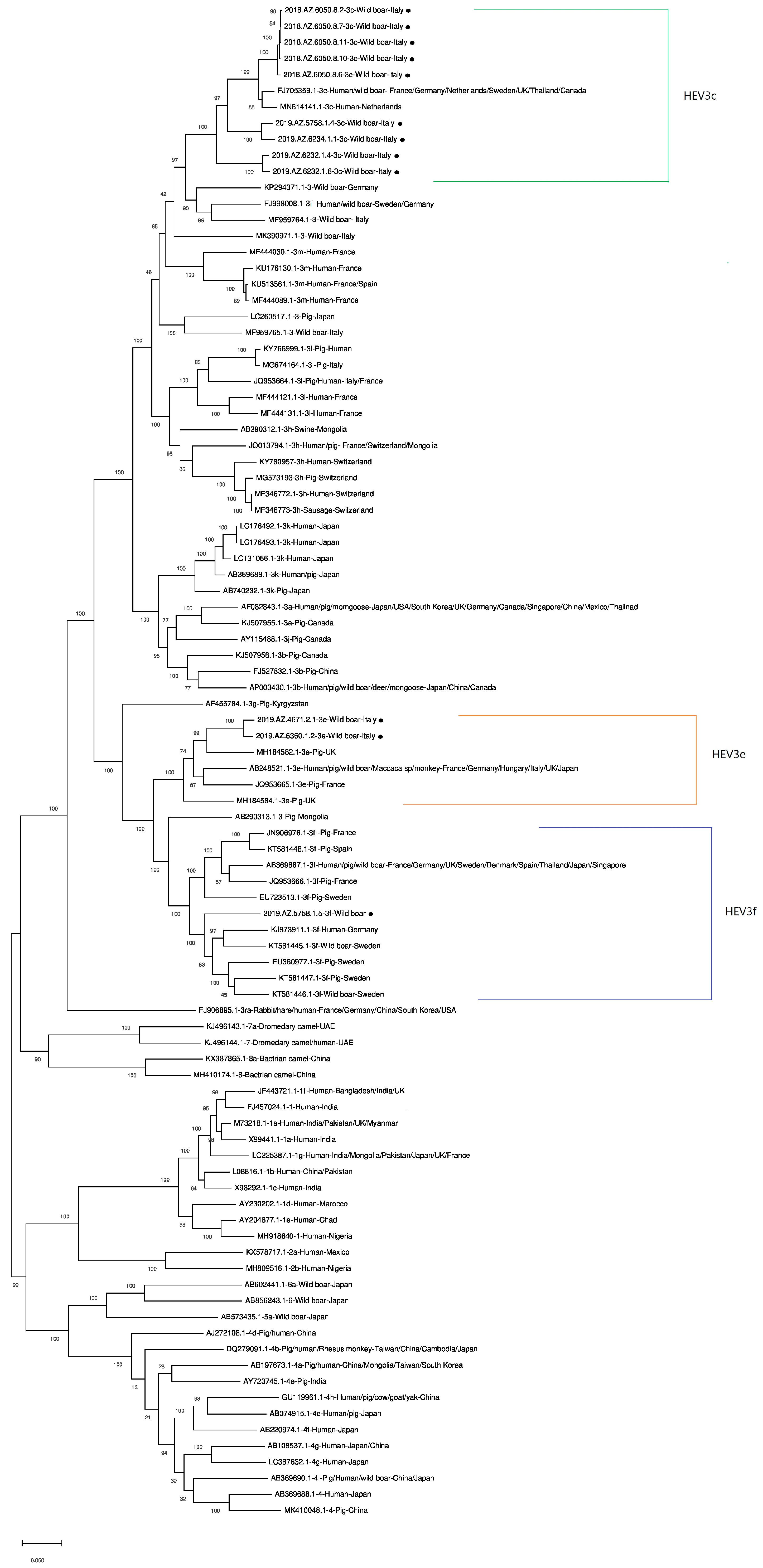
2.4. Phylogenetic Analysis
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- EFSA Panel on Biological, Hazards; Ricci, A.; Allende, A.; Bolton, D.; Chemaly, M.; Davies, R.; Escamez, P.S.F.; Herman, L.; Koutsoumanis, K.; Lindqvist, R.; et al. Public health risks associated with hepatitis E virus (HEV) as a food-borne pathogen. EFSA J. 2017, 15, 04886. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Gracia, M.T.; Suay-García, B.; Mateos-Lindemann, M.L. Hepatitis E and pregnancy: Current state. Rev. Med. Virol. 2017, 27, e1929. [Google Scholar] [CrossRef] [PubMed]
- Harritshoej, L.H.; Hother, C.E.; Sengeløv, H.; Daugaard, G.; Sørensen, S.S.; Jacobsen, S.; Perch, M.; Holm, D.K.; Sækmose, S.G.; Aagaard, B.; et al. Epidemiology of hepatitis E virus infection in a cohort of 4023 immunocompromised patients. Int. J. Infect. Dis. 2019, 91, 188–195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woo, P.C.Y.; Lau, S.K.P.; Teng, J.L.; Cao, K.-Y.; Wernery, U.; Schountz, T.; Chiu, T.H.; Tsang, A.K.; Wong, P.-C.; Wong, E.Y.; et al. New Hepatitis E Virus Genotype in Bactrian Camels, Xinjiang, China, 2013. Emerg. Infect. Dis. 2016, 22, 2219–2221. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.B.; Izopet, J.; Nicot, F.; Simmonds, P.; Jameel, S.; Meng, X.-J.; Norder, H.; Okamoto, H.; Van Der Poel, W.H.; Reuter, G.; et al. Update: Proposed reference sequences for subtypes of hepatitis E virus (species Orthohepevirus A). J. Gen. Virol. 2020, 101, 692–698. [Google Scholar] [CrossRef] [PubMed]
- Kamar, N.; Izopet, J.; Rostaing, L. Hepatitis E virus infection. Curr. Opin. Gastroenterol. 2013, 29, 271–278. [Google Scholar] [CrossRef]
- Aspinall, E.J.; Couturier, E.; Faber, M.; Said, B.; Ijaz, S.; Tavoschi, L.; Takkinen, J.; Adlhoch, C. Hepatitis E virus infection in Europe: Surveillance and descriptive epidemiology of confirmed cases, 2005 to 2015. Eurosurveillance 2017, 22, 30561. [Google Scholar] [CrossRef] [Green Version]
- Fredriksson-Ahomaa, M. Wild Boar: A Reservoir of Foodborne Zoonoses. Foodborne Pathog. Dis. 2019, 16, 153–165. [Google Scholar] [CrossRef]
- Alfonsi, V.; Romanò, L.; Ciccaglione, A.R.; La Rosa, G.; Bruni, R.; Zanetti, A.; Della Libera, S.; Iaconelli, M.; Bagnarelli, P.; Capobianchi, M.R.; et al. Hepatitis E in Italy: 5 years of national epidemiological, virological and environmental surveillance, 2012 to 2016. Eurosurveillance 2018, 23, 1700517. [Google Scholar] [CrossRef]
- Caruso, C.; Modesto, P.; Bertolini, S.; Peletto, S.; Acutis, P.L.; Dondo, A.; Robetto, S.; Mignone, W.; Orusa, R.; Ru, G.; et al. Serological and virological survey of hepatitis E virus in wild boar populations in northwestern Italy: Detection of HEV subtypes 3e and 3f. Arch. Virol. 2014, 160, 153–160. [Google Scholar] [CrossRef]
- Di Profio, F.; Melegari, I.; Sarchese, V.; Robetto, S.; Marruchella, G.; Bona, M.C.; Orusa, R.; Martella, V.; Marsilio, F.; Di Martino, B. Detection and genetic characterization of hepatitis E virus (HEV) genotype 3 subtype c in wild boars in Italy. Arch. Virol. 2016, 161, 2829–2834. [Google Scholar] [CrossRef] [PubMed]
- Di Pasquale, S.; De Santis, P.; La Rosa, G.; Di Domenico, K.; Iaconelli, M.; Micarelli, G.; Martini, E.; Bilei, S.; De Medici, D.; Suffredini, E. Quantification and genetic diversity of Hepatitis E virus in wild boar (Sus scrofa) hunted for domestic consumption in Central Italy. Food Microbiol. 2019, 82, 194–201. [Google Scholar] [CrossRef] [PubMed]
- De Sabato, L.; Lemey, P.; Vrancken, B.; Bonfanti, L.; Ceglie, L.; Vaccari, G.; Di Bartolo, I. Proposal for a new subtype of the zoonotic genotype 3 Hepatitis E virus: HEV-3l. Virus Res. 2018, 248, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Aprea, G.; Amoroso, M.G.; Di Bartolo, I.; D’Alessio, N.; Di Sabatino, D.; Boni, A.; Cioffi, B.; D’Angelantonio, D.; Scattolini, S.; De Sabato, L.; et al. Molecular detection and phylogenetic analysis of hepatitis E virus strains circulating in wild boars in south-central Italy. Transbound. Emerg. Dis. 2017, 65, 813–e31. [Google Scholar] [CrossRef]
- Lucarelli, C.; Spada, E.; Taliani, G.; Chionne, P.; Madonna, E.; Marcantonio, C.; Pezzotti, P.; Bruni, R.; La Rosa, G.; Pisani, G.; et al. High prevalence of anti-hepatitis E virus antibodies among blood donors in central Italy, February to March 2014. Eurosurveillance 2016, 21, 30299. [Google Scholar] [CrossRef]
- Sarchese, V.; Di Profio, F.; Melegari, I.; Palombieri, A.; Sanchez, S.B.; Arbuatti, A.; Ciuffetelli, M.; Marsilio, F.; Martella, V.; Di Martino, B. Hepatitis E virus in sheep in Italy. Transbound. Emerg. Dis. 2019, 66, 1120–1125. [Google Scholar] [CrossRef]
- Di Profio, F.; Melegari, I.; Palombieri, A.; Sarchese, V.; Arbuatti, A.; Fruci, P.; Marsilio, F.; Martella, V.; Di Martino, B. High prevalence of hepatitis E virus in raw sewage in Southern Italy. Virus Res. 2019, 272, 19771. [Google Scholar] [CrossRef]
- De Sabato, L.; Ostanello, F.; De Grossi, L.; Marcario, A.; Franzetti, B.; Monini, M.; Di Bartolo, I. Molecular survey of HEV infection in wild boar population in Italy. Transbound. Emerg. Dis. 2018, 65, 1749–1756. [Google Scholar] [CrossRef]
- Montone, A.M.I.; De Sabato, L.; Suffredini, E.; Alise, M.; Zaccherini, A.; Volzone, P.; Di Maro, O.; Neola, B.; Capuano, F.; Di Bartolo, I. Occurrence of HEV-RNA in Italian Regional Pork and Wild Boar Food Products. Food Environ. Virol. 2019, 11, 420–426. [Google Scholar] [CrossRef]
- Smith, D.B.; Paddy, J.O.; Simmonds, P. The use of human sewage screening for community surveillance of hepatitis E virus in the UK. J. Med. Virol. 2015, 88, 915–918. [Google Scholar] [CrossRef] [Green Version]
- Vina-Rodríguez, A.; Schlosser, J.; Becher, D.; Kaden, V.; Groschup, M.H.; Eiden, M. Hepatitis E Virus Genotype 3 Diversity: Phylogenetic Analysis and Presence of Subtype 3b in Wild Boar in Europe. Viruses 2015, 7, 2704–2726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Bartolo, I.; Diez-Valcarce, M.; Vasickova, P.; Kralik, P.; Hernández, M.; Angeloni, G.; Ostanello, F.; Bouwknegt, M.; Rodríguez-Lázaro, D.; Pavlik, I.; et al. Hepatitis E Virus in Pork Production Chain in Czech Republic, Italy, and Spain, 2010. Emerg. Infect. Dis. 2012, 18, 1282–1289. [Google Scholar] [CrossRef] [PubMed]
- Jothikumar, N.; Cromeans, T.L.; Robertson, B.H.; Meng, X.; Hill, V.R. A broadly reactive one-step real-time RT-PCR assay for rapid and sensitive detection of hepatitis E virus. J. Virol. Methods 2006, 131, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Martinez, M.; Diez-Valcarce, M.; Hernández, M.; Rodríguez-Lázaro, D. Design and Application of Nucleic Acid Standards for Quantitative Detection of Enteric Viruses by Real-Time PCR. Food Environ. Virol. 2011, 3, 92–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diez-Valcarce, M.; Cook, N.; Hernández, M.; Rodríguez-Lázaro, D. Analytical Application of a Sample Process Control in Detection of Foodborne Viruses. Food Anal. Methods 2011, 4, 614–618. [Google Scholar] [CrossRef] [Green Version]
- Lorusso, A.; Calistri, P.; Mercante, M.T.; Monaco, F.; Portanti, O.; Marcacci, M.; Cammà, C.; Rinaldi, A.; Mangone, I.; Di Pasquale, A.; et al. A “One-Health” approach for diagnosis and molecular characterization of SARS-CoV-2 in Italy. One Health 2020, 10, 100135. [Google Scholar] [CrossRef]
- Cito, F.; Di Pasquale, A.; Cammà, C.; Cito, P. The Italian information system for the collection and analysis of complete genome sequence of pathogens isolated from animal, food and environment. Int. J. Infect. Dis. 2018, 73, 296–297. [Google Scholar] [CrossRef]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [Green Version]
- Seemann, T. ABRicate: Mass Screening of Contigs for Antimicrobial and Virulence Genes; Department of Microbiology and Immunology, The University of Melbourne: Melbourne, Australia. Available online: https://github.com/tseemann/abricate (accessed on 28 February 2019).
- Baker, M. Ben Langmead: Building a better sequence alignment program. Nat. Methods 2012, 9, 313–314. [Google Scholar] [CrossRef]
- Tamura, K.; Nei, M. Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol. Biol. Evol. 1993, 10, 512–526. [Google Scholar]
- Mulder, A.C.; Kroneman, A.; Franz, E.; Vennema, H.; Tulen, A.D.; Takkinen, J.; Hofhuis, A.; Adlhoch, C. HEVnet: A One Health, collaborative, interdisciplinary network and sequence data repository for enhanced hepatitis E virus molecular typing, characterisation and epidemiological investigations. Eurosurveillance 2019, 24, 1800407. [Google Scholar] [CrossRef] [PubMed]
- Monini, M.; Di Bartolo, I.; Ianiro, G.; Angeloni, G.; Magistrali, C.F.; Ostanello, F.; Ruggeri, F.M. Detection and molecular characterization of zoonotic viruses in swine fecal samples in Italian pig herds. Arch. Virol. 2015, 160, 2547–2556. [Google Scholar] [CrossRef]
- Schielke, A.; Sachs, K.; Lierz, M.; Appel, B.; Jansen, A.; Johne, R. Detection of hepatitis E virus in wild boars of rural and urban regions in Germany and whole genome characterization of an endemic strain. Virol. J. 2009, 6, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Sari, G.; van de Garde, M.D.B.; van Schoonhoven, A.; Voermans, J.J.C.; van der Eijk, A.A.; de Man, R.A.; Boonstra, A.; Vanwolleghem, T.; Pas, S.D. Hepatitis e virus shows more genomic alterations in cell culture than in vivo. Pathogens 2019, 8, 255. [Google Scholar] [CrossRef] [Green Version]
- Bouquet, J.; Cherel, P.; Pavio, N. Genetic characterization and codon usage bias of full-length Hepatitis E virus sequences shed new lights on genotypic distribution, host restriction and genome evolution. Infect. Genet. Evol. 2012, 12, 1842–1853. [Google Scholar] [CrossRef]
- Grierson, S.S.; McGowan, S.; Cook, C.; Steinbach, F.; Choudhury, B. Molecular and in vitro characterisation of hepatitis E virus from UK pigs. Virology 2019, 527, 116–121. [Google Scholar] [CrossRef] [PubMed]
- Huzly, D.; Umhau, M.; Bettinger, D.; Cathomen, T.; Emmerich, F.; Hasselblatt, P.; Hengel, H.; Herzog, R.; Kappert, O.; Maassen, S. Transfusion-transmitted hepatitis E in Germany, 2013. Eurosurveillance 2014, 19, 20812. [Google Scholar] [CrossRef] [PubMed]
- Xia, H.; Liu, L.; Linde, A.-M.; Belák, S.; Norder, H.; Widén, F. Molecular characterization and phylogenetic analysis of the complete genome of a hepatitis E virus from European swine. Virus Genes 2008, 37, 39–48. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| ID | Hunting Area | Year | Age | Sex | HEV Subtype |
|---|---|---|---|---|---|
| 2018-AZ-6050-8-2 | L’Aquila | 2018 | 6 months | Female | HEV3-c |
| 2018-AZ-6050-8-6 | L’Aquila | 2018 | 6 months | Female | HEV3-c |
| 2018-AZ-6050-8-7 | L’Aquila | 2018 | 6 months | Female | HEV3-c |
| 2018-AZ-6050-8-10 | L’Aquila | 2018 | 6 months | Female | HEV3-c |
| 2018-AZ-6050-8-11 | L’Aquila | 2018 | 10 months | Female | HEV3-c |
| 2019-AZ-6360-1-2 | Teramo | 2019 | 12 months | Female | HEV3-e |
| 2019-AZ-6234-1-1 | L’Aquila | 2019 | 6 months | Male | HEV3-c |
| 2019-AZ-6232-1-6 | L’Aquila | 2019 | - | Female | HEV3-c |
| 2019-AZ-6232-1-4 | L’Aquila | 2019 | - | Female | HEV3-c |
| 2019-AZ-5758-1-5 | Pescara | 2019 | - | HEV3 f | |
| 2019-AZ-5758-1-4 | Pescara | 2019 | - | HEV3-c | |
| 2019-AZ-4671-2-1 | L’Aquila | 2019 | 6 months | Male | HEV3-e |
| Sample_ID | Total Rawreads | Q30 Rawreads | Total Trimreads | Q30 Trimreads | Sequence Lenght | %GC | Coverage |
|---|---|---|---|---|---|---|---|
| 2018.AZ.6050.8.2 | 5,070,316 | 84.71 | 3,686,844 | 92.72 | 7210 | 55.5 | 775.17 |
| 2018.AZ.6050.8.6 | 20,142 | 85.72 | 14,560 | 93.05 | 6736 | 55.4 | 5.87 |
| 2018.AZ.6050.8.7 | 2,483,214 | 84.95 | 1,705,664 | 93.09 | 7067 | 55.7 | 369.24 |
| 2018.AZ.6050.8.10 | 1,560,834 | 84.63 | 1,114,066 | 92.82 | 7006 | 55.6 | 88.12 |
| 2018.AZ.6050.8.11 | 924,066 | 85.86 | 832,502 | 93.11 | 7039 | 55.7 | 63.06 |
| 2019.AZ.4671.2.1 | 458,770 | 95.47 | 440,458 | 97.71 | 7237 | 55.1 | 1678.89 |
| 2019.AZ.5758.1.4 | 1,568,372 | 86.03 | 1,469,764 | 92.5 | 7107 | 56 | 54.62 |
| 2019.AZ.5758.1.5 | 767,482 | 93.91 | 730,209 | 97.93 | 7024 | 55.7 | 8.39 |
| 2019.AZ.6232.1.4 | 1,224,774 | 84.32 | 1,124,515 | 93.19 | 7042 | 55.3 | 8.58 |
| 2019.AZ.6232.1.6 | 488,730 | 92.35 | 451,677 | 97.72 | 7206 | 55.4 | 7.49 |
| 2019.AZ.6234.1.1 | 844,282 | 94.76 | 806,030 | 97.95 | 7087 | 55.6 | 174.86 |
| 2019.AZ.6360.1.2 | 444,820 | 90.58 | 423,140 | 97.63 | 7191 | 55.2 | 130.17 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aprea, G.; Scattolini, S.; D’Angelantonio, D.; Chiaverini, A.; Di Lollo, V.; Olivieri, S.; Marcacci, M.; Mangone, I.; Salucci, S.; Antoci, S.; et al. Whole Genome Sequencing Characterization of HEV3-e and HEV3-f Subtypes among the Wild Boar Population in the Abruzzo Region, Italy: First Report. Microorganisms 2020, 8, 1393. https://doi.org/10.3390/microorganisms8091393
Aprea G, Scattolini S, D’Angelantonio D, Chiaverini A, Di Lollo V, Olivieri S, Marcacci M, Mangone I, Salucci S, Antoci S, et al. Whole Genome Sequencing Characterization of HEV3-e and HEV3-f Subtypes among the Wild Boar Population in the Abruzzo Region, Italy: First Report. Microorganisms. 2020; 8(9):1393. https://doi.org/10.3390/microorganisms8091393
Chicago/Turabian StyleAprea, Giuseppe, Silvia Scattolini, Daniela D’Angelantonio, Alexandra Chiaverini, Valeria Di Lollo, Sabrina Olivieri, Maurilia Marcacci, Iolanda Mangone, Stefania Salucci, Salvatore Antoci, and et al. 2020. "Whole Genome Sequencing Characterization of HEV3-e and HEV3-f Subtypes among the Wild Boar Population in the Abruzzo Region, Italy: First Report" Microorganisms 8, no. 9: 1393. https://doi.org/10.3390/microorganisms8091393