Equine Alphaherpesviruses Require Activation of the Small GTPases Rac1 and Cdc42 for Intracellular Transport


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells and Viruses
2.2. Inhibitors
2.3. Cytotoxicity Assay
2.4. Flow Cytometry
2.5. Plaque Assay
2.6. Virus Localization and Immunofluorescence
2.7. Ratiometric Fluorescence Resonance Energy Transfer (FRET)
2.8. Immunoblotting
2.9. Cell-to-Cell Fusion
2.10. Statistical Analysis
3. Results
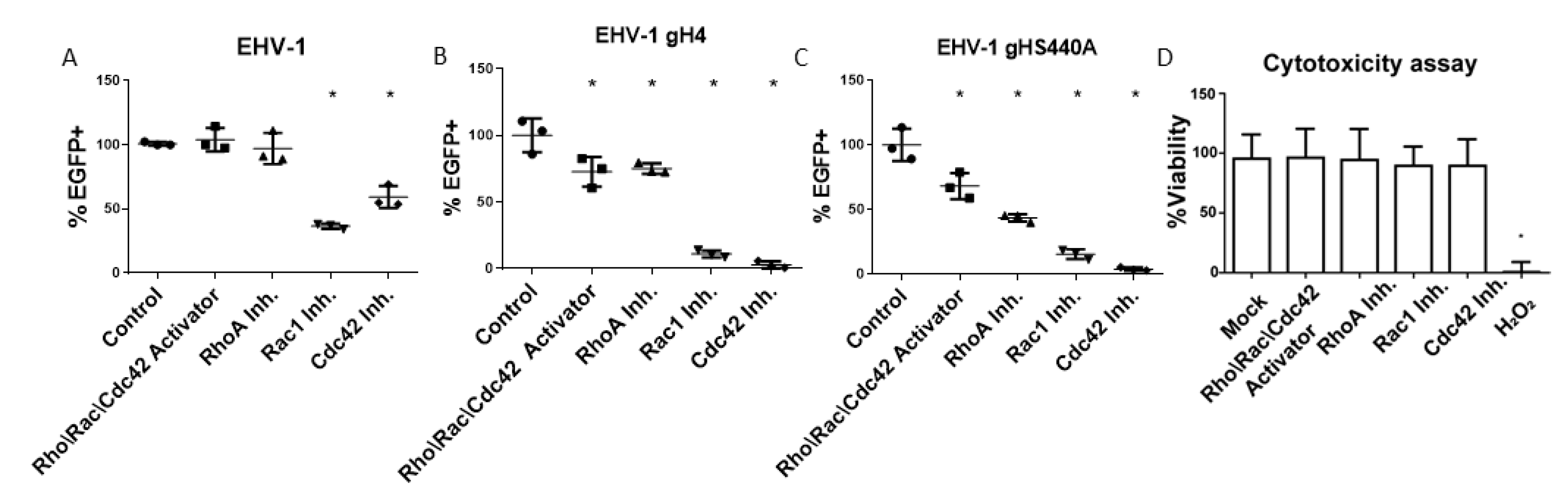
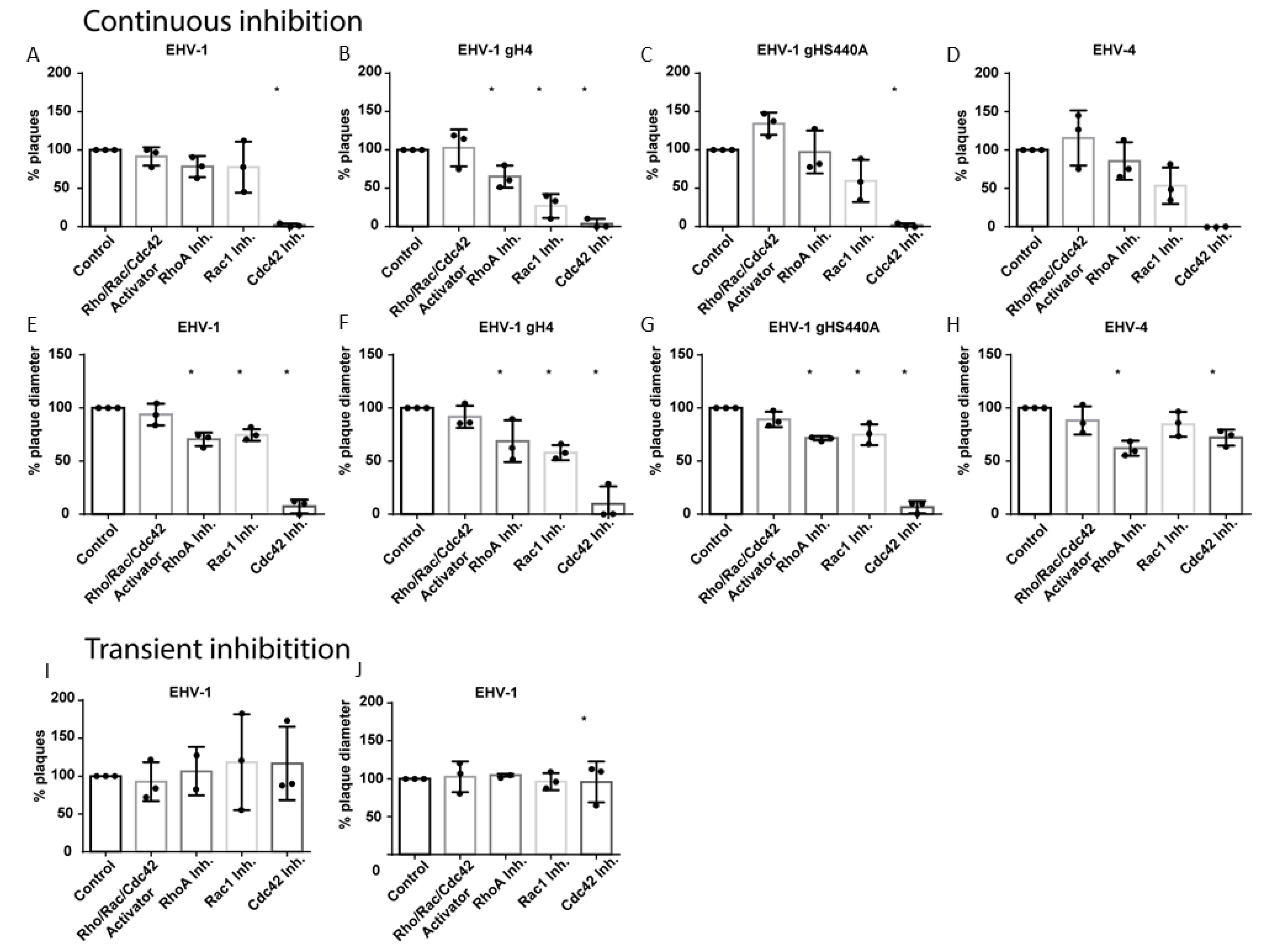
3.1. Cdc42 and Rac1 Inhibitors Reduce EHV-1 and EHV-4 Infection
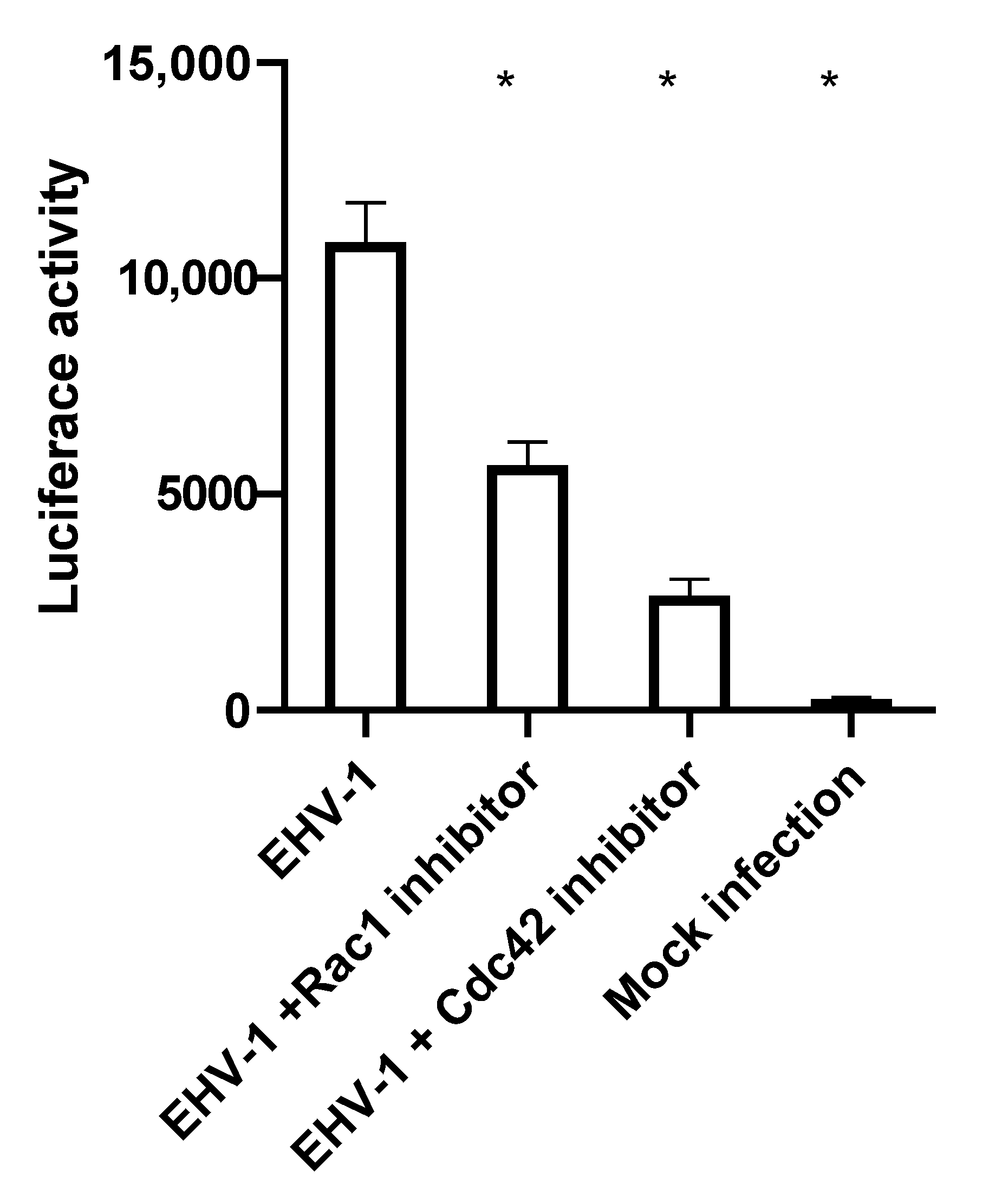
3.2. Small GTPases Facilitate Infection and Cell-to-Cell Spread
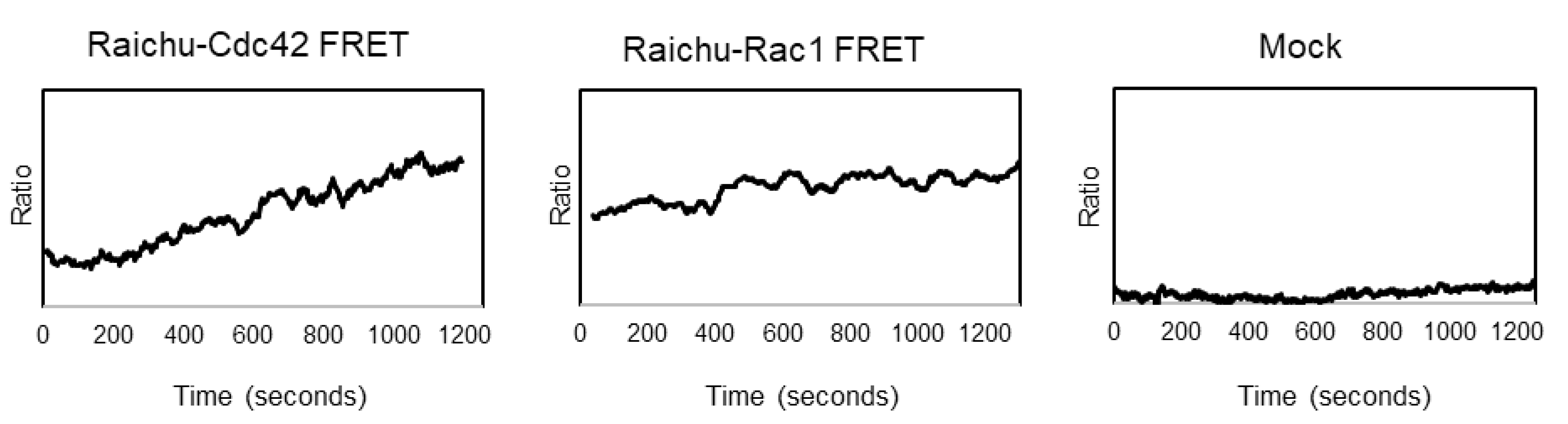
3.3. EHV-1 Activates Small GTPases Rac1 and Cdc42
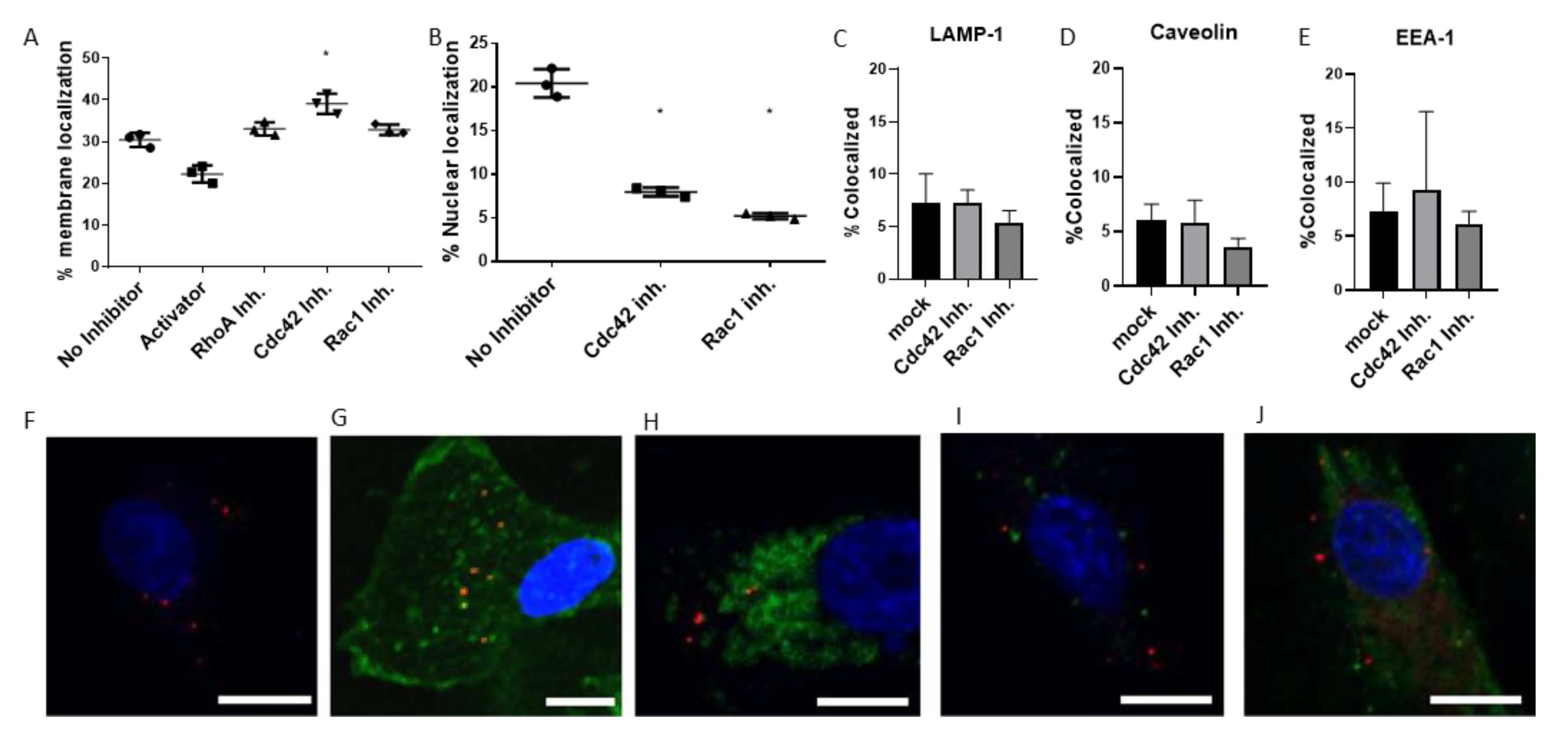
3.4. Tracking of Virus Transport in Cells
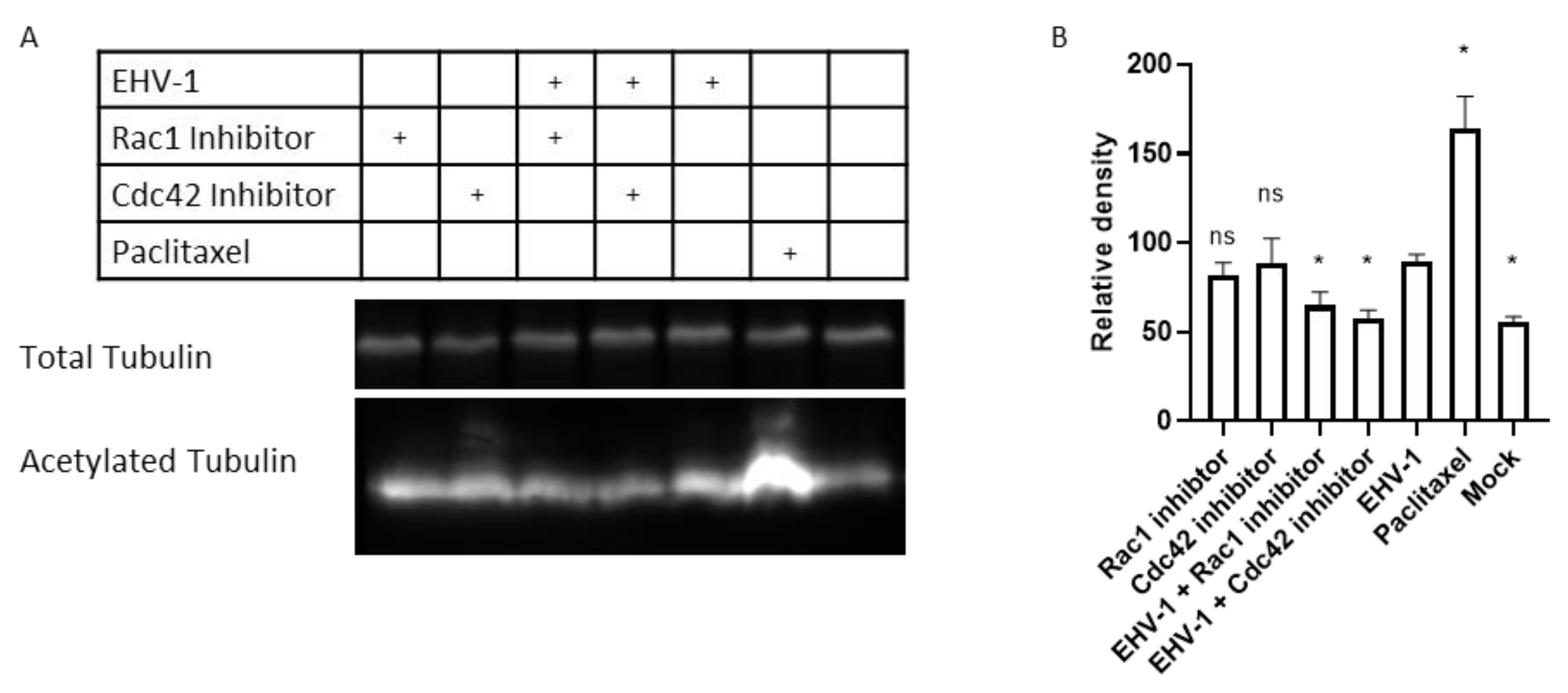
3.5. Rac1 and Cdc42 Activation Is Required for EHV-1-Induced Tubulin Acetylation
3.6. EHV-1-Induced Cell-to-Cell Fusion Is Dependent on Rac1 and Cdc42 Activation
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Alberts, B.; Johnson, A.; Lewis, J.; Raff, M.; Roberts, K.; Walter, P. Molecular Biology of the Cell, 4th ed.; Garland Science: New York, NY, USA, 2002; ISBN 978-0-8153-3218-3. [Google Scholar]
- Haga, R.B.; Ridley, A.J. Rho GTPases: Regulation and roles in cancer cell biology. Small GTPases 2016, 7, 207–221. [Google Scholar] [CrossRef] [Green Version]
- Hall, A. Rho GTPases and the control of cell behaviour. Biochem. Soc. Trans. 2005, 33, 891–895. [Google Scholar] [CrossRef] [PubMed]
- Kötting, C.; Gerwert, K. The dynamics of the catalytic site in small GTPases, variations on a common motif. FEBS Lett. 2013, 587, 2025–2027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aslan, J.E.; McCarty, O.J.T. Rho GTPases in Platelet Function. J. Thromb. Haemost. JTH 2013, 11, 35–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nobes, C.D.; Hall, A. Rho GTPases Control Polarity, Protrusion, and Adhesion during Cell Movement. J. Cell Biol. 1999, 144, 1235–1244. [Google Scholar] [CrossRef] [Green Version]
- Madaule, P.; Axel, R. A novel ras-related gene family. Cell 1985, 41, 31–40. [Google Scholar] [CrossRef]
- Boettner, B.; Van Aelst, L. The role of Rho GTPases in disease development. Gene 2002, 286, 155–174. [Google Scholar] [CrossRef]
- Ellenbroek, S.I.J.; Collard, J.G. Rho GTPases: Functions and association with cancer. Clin. Exp. Metastasis 2007, 24, 657–672. [Google Scholar] [CrossRef]
- Fujioka, Y.; Tsuda, M.; Nanbo, A.; Hattori, T.; Sasaki, J.; Sasaki, T.; Miyazaki, T.; Ohba, Y. A Ca2+-dependent signalling circuit regulates influenza A virus internalization and infection. Nat. Commun. 2013, 4, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.-L.; Zhang, J.-L.; Chen, W.; Xu, X.-F.; Gao, N.; Fan, D.-Y.; An, J. Roles of Small GTPase Rac1 in the Regulation of Actin Cytoskeleton during Dengue Virus Infection. PLoS Negl. Trop. Dis. 2010, 4, e809. [Google Scholar] [CrossRef] [Green Version]
- Hoppe, S.; Schelhaas, M.; Jaeger, V.; Liebig, T.; Petermann, P.; Knebel-Mörsdorf, D. Early herpes simplex virus type 1 infection is dependent on regulated Rac1/Cdc42 signalling in epithelial MDCKII cells. J. Gen. Virol. 2006, 87, 3483–3494. [Google Scholar] [CrossRef] [PubMed]
- Kurtz, B.M.; Singletary, L.B.; Kelly, S.D.; Frampton, A.R. Equus caballus Major Histocompatibility Complex Class I Is an Entry Receptor for Equine Herpesvirus Type 1. J. Virol. 2010, 84, 9027–9034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azab, W.; Osterrieder, N. Glycoproteins D of Equine Herpesvirus Type 1 (EHV-1) and EHV-4 Determine Cellular Tropism Independently of Integrins. J. Virol. 2012, 86, 2031–2044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azab, W.; Gramatica, A.; Herrmann, A.; Osterrieder, N. Binding of Alphaherpesvirus Glycoprotein H to Surface α4β1-Integrins Activates Calcium-Signaling Pathways and Induces Phosphatidylserine Exposure on the Plasma Membrane. mBio 2015, 6, e01552-15. [Google Scholar] [CrossRef] [Green Version]
- Price, L.S.; Langeslag, M.; ten Klooster, J.P.; Hordijk, P.L.; Jalink, K.; Collard, J.G. Calcium signaling regulates translocation and activation of Rac. J. Biol. Chem. 2003, 278, 39413–39421. [Google Scholar] [CrossRef] [Green Version]
- Saneyoshi, T.; Hayashi, Y. The Ca2+ and Rho GTPase signaling pathways underlying activity-dependent actin remodeling at dendritic spines. Cytoskelet. Hoboken NJ 2012, 69, 545–554. [Google Scholar] [CrossRef]
- Wilk-Blaszczak, M.A.; Singer, W.D.; Quill, T.; Miller, B.; Frost, J.A.; Sternweis, P.C.; Belardetti, F. The monomeric G-proteins Rac1 and/or Cdc42 are required for the inhibition of voltage-dependent calcium current by bradykinin. J. Neurosci. Off. J. Soc. Neurosci. 1997, 17, 4094–4100. [Google Scholar] [CrossRef]
- Azab, W.; Zajic, L.; Osterrieder, N. The role of glycoprotein H of equine herpesviruses 1 and 4 (EHV-1 and EHV-4) in cellular host range and integrin binding. Vet. Res. 2012, 43, 61. [Google Scholar] [CrossRef] [Green Version]
- Azab, W.; Kato, K.; Arii, J.; Tsujimura, K.; Yamane, D.; Tohya, Y.; Matsumura, T.; Akashi, H. Cloning of the genome of equine herpesvirus 4 strain TH20p as an infectious bacterial artificial chromosome. Arch. Virol. 2009, 154, 833–842. [Google Scholar] [CrossRef]
- Rauth, A.M. The Physical State of Viral Nucleic Acid and the Sensitivity of Viruses to Ultraviolet Light. Biophys. J. 1965, 5, 257–273. [Google Scholar] [CrossRef] [Green Version]
- Surviladze, Z.; Waller, A.; Strouse, J.J.; Bologa, C.; Ursu, O.; Salas, V.; Parkinson, J.F.; Phillips, G.K.; Romero, E.; Wandinger-Ness, A.; et al. A Potent and Selective Inhibitor of Cdc42 GTPase. In Probe Reports from the NIH Molecular Libraries Program; National Center for Biotechnology Information (US): Bethesda, MD, USA, 2010. [Google Scholar]
- Aguilar, J.; Roy, D.; Ghazal, P.; Wagner, E. Dimethyl sulfoxide blocks herpes simplex virus-1 productive infection in vitro acting at different stages with positive cooperativity. Application of micro-array analysis. BMC Infect. Dis. 2002, 2, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ngamwongsatit, P.; Banada, P.P.; Panbangred, W.; Bhunia, A.K. WST-1-based cell cytotoxicity assay as a substitute for MTT-based assay for rapid detection of toxigenic Bacillus species using CHO cell line. J. Microbiol. Methods 2008, 73, 211–215. [Google Scholar] [CrossRef] [PubMed]
- Dey, P.; Bergmann, T.; Cuellar-Camacho, J.L.; Ehrmann, S.; Chowdhury, M.S.; Zhang, M.; Dahmani, I.; Haag, R.; Azab, W. Multivalent Flexible Nanogels Exhibit Broad-Spectrum Antiviral Activity by Blocking Virus Entry. ACS Nano 2018, 12, 6429–6442. [Google Scholar] [CrossRef] [PubMed]
- Mochizuki, N.; Yamashita, S.; Kurokawa, K.; Ohba, Y.; Nagai, T.; Miyawaki, A.; Matsuda, M. Spatio-temporal images of growth-factor-induced activation of Ras and Rap1. Nature 2001, 411, 1065–1068. [Google Scholar] [CrossRef] [PubMed]
- Yoshizaki, H.; Ohba, Y.; Kurokawa, K.; Itoh, R.E.; Nakamura, T.; Mochizuki, N.; Nagashima, K.; Matsuda, M. Activity of Rho-family GTPases during cell division as visualized with FRET-based probes. J. Cell Biol. 2003, 162, 223–232. [Google Scholar] [CrossRef] [PubMed]
- Frampton, A.R.; Uchida, H.; von Einem, J.; Goins, W.F.; Grandi, P.; Cohen, J.B.; Osterrieder, N.; Glorioso, J.C. Equine herpesvirus type 1 (EHV-1) utilizes microtubules, dynein, and ROCK1 to productively infect cells. Vet. Microbiol. 2010, 141, 12–21. [Google Scholar] [CrossRef] [Green Version]
- Ji, H. Lysis of Cultured Cells for Immunoprecipitation. Cold Spring Harb. Protoc. 2010, 8, pdb-prot5466. [Google Scholar] [CrossRef]
- Westermeier, R. Method 9: Semi-Dry Blotting of Proteins. In Electrophoresis in Practice; John Wiley & Sons Ltd.: London, UK, 2005; pp. 247–255. ISBN 978-3-527-60346-6. [Google Scholar]
- Using ImageJ to Quantify Blots. Available online: https://di.uq.edu.au/community-and-alumni/sparq-ed/sparq-ed-services/using-imagej-quantify-blots (accessed on 11 March 2020).
- Sorem, J.; Longnecker, R. Cleavage of Epstein-Barr Virus Glycoprotein B is required for Full Function in Cell:Cell Fusion with both Epithelial and B Cells. J. Gen. Virol. 2009, 90, 591–595. [Google Scholar] [CrossRef]
- Distler, J.H.W.; Jüngel, A.; Kurowska-Stolarska, M.; Michel, B.A.; Gay, R.E.; Gay, S.; Distler, O. Nucleofection: A new, highly efficient transfection method for primary human keratinocytes*. Exp. Dermatol. 2005, 14, 315–320. [Google Scholar] [CrossRef]
- Fleming, I.N.; Elliott, C.M.; Buchanan, F.G.; Downes, C.P.; Exton, J.H. Ca2+/calmodulin-dependent protein kinase II regulates Tiam1 by reversible protein phosphorylation. J. Biol. Chem. 1999, 274, 12753–12758. [Google Scholar] [CrossRef] [Green Version]
- Lyman, M.G.; Enquist, L.W. Herpesvirus Interactions with the Host Cytoskeleton. J. Virol. 2009, 83, 2058–2066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sodeik, B.; Ebersold, M.W.; Helenius, A. Microtubule-mediated transport of incoming herpes simplex virus 1 capsids to the nucleus. J. Cell Biol. 1997, 136, 1007–1021. [Google Scholar] [CrossRef] [PubMed]
- Schulze, E.; Asai, D.J.; Bulinski, J.C.; Kirschner, M. Posttranslational modification and microtubule stability. J. Cell Biol. 1987, 105, 2167–2177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takemura, R.; Okabe, S.; Umeyama, T.; Kanai, Y.; Cowan, N.J.; Hirokawa, N. Increased microtubule stability and alpha tubulin acetylation in cells transfected with microtubule-associated proteins MAP1B, MAP2 or tau. J. Cell Sci. 1992, 103, 953–964. [Google Scholar] [PubMed]
- Bulinski, J.C.; Richards, J.E.; Piperno, G. Posttranslational modifications of alpha tubulin: Detyrosination and acetylation differentiate populations of interphase microtubules in cultured cells. J. Cell Biol. 1988, 106, 1213–1220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reed, N.A.; Cai, D.; Blasius, T.L.; Jih, G.T.; Meyhofer, E.; Gaertig, J.; Verhey, K.J. Microtubule Acetylation Promotes Kinesin-1 Binding and Transport. Curr. Biol. 2006, 16, 2166–2172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, M.; Zheng, K.; Chen, M.; Xiang, Y.; Jin, F.; Ma, K.; Qiu, X.; Wang, Q.; Peng, T.; Kitazato, K.; et al. Heat-Shock Protein 90 Promotes Nuclear Transport of Herpes Simplex Virus 1 Capsid Protein by Interacting with Acetylated Tubulin. PLoS ONE 2014, 9, e99425. [Google Scholar] [CrossRef] [Green Version]
- Turner, A.; Bruun, B.; Minson, T.; Browne, H. Glycoproteins gB, gD, and gHgL of Herpes Simplex Virus Type 1 Are Necessary and Sufficient to Mediate Membrane Fusion in a Cos Cell Transfection System. J. Virol. 1998, 72, 873–875. [Google Scholar] [CrossRef] [Green Version]
- Wittmann, T.; Waterman-Storer, C.M. Cell motility: Can Rho GTPases and microtubules point the way? J. Cell Sci. 2001, 114, 3795–3803. [Google Scholar]
- Csellner, H.; Walker, C.; Wellington, J.E.; McLure, L.E.; Love, D.N.; Whalley, J.M. EHV-1 glycoprotein D (EHV-1 gD) is required for virus entry and cell-cell fusion, and an EHV-1 gD deletion mutant induces a protective immune response in mice. Arch. Virol. 2000, 145, 2371–2385. [Google Scholar] [CrossRef] [PubMed]
- Zamudio-Meza, H.; Castillo-Alvarez, A.; González-Bonilla, C.; Meza, I. Cross-talk between Rac1 and Cdc42 GTPases regulates formation of filopodia required for dengue virus type-2 entry into HMEC-1 cells. J. Gen. Virol. 2009, 90, 2902–2911. [Google Scholar] [CrossRef] [PubMed]
- Lv, C.; Lin, Y.; Sun, E.-Z.; Tang, B.; Ao, J.; Wang, J.-J.; Zhang, Z.-L.; Zheng, Z.; Wang, H.; Pang, D.-W. Internalization of the pseudorabies virus via macropinocytosis analyzed by quantum dot-based single-virus tracking. Chem. Commun. 2018, 54, 11184–11187. [Google Scholar] [CrossRef] [PubMed]
- Petermann, P.; Haase, I.; Knebel-Mörsdorf, D. Impact of Rac1 and Cdc42 Signaling during Early Herpes Simplex Virus Type 1 Infection of Keratinocytes. J. Virol. 2009, 83, 9759–9772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Donnell, C.D.; Shukla, D. A novel function of heparan sulfate in the regulation of cell-cell fusion. J. Biol. Chem. 2009, 284, 29654–29665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eitzen, G. Actin remodeling to facilitate membrane fusion. Biochim. Biophys. Acta BBA Mol. Cell Res. 2003, 1641, 175–181. [Google Scholar] [CrossRef] [Green Version]
- Ellis, S.; Mellor, H. The novel Rho-family GTPase Rif regulates coordinated actin-based membrane rearrangements. Curr. Biol. 2000, 10, 1387–1390. [Google Scholar] [CrossRef] [Green Version]
- Azab, W.; Lehmann, M.J.; Osterrieder, N. Glycoprotein H and α4β1 integrins determine the entry pathway of alphaherpesviruses. J. Virol. 2013, 87, 5937–5948. [Google Scholar] [CrossRef] [Green Version]
- Reszka, N.; Zhou, C.; Song, B.; Sodroski, J.G.; Knipe, D.M. Simian TRIM5α Proteins Reduce Replication of Herpes Simplex Virus. Virology 2010, 398, 243–250. [Google Scholar] [CrossRef] [Green Version]
- Cohen, E.M.; Avital, N.; Shamay, M.; Kobiler, O. Abortive herpes simplex virus infection of nonneuronal cells results in quiescent viral genomes that can reactivate. Proc. Natl. Acad. Sci. USA 2020, 117, 635–640. [Google Scholar] [CrossRef]
- Wu, W.J.; Erickson, J.W.; Lin, R.; Cerione, R.A. The γ-subunit of the coatomer complex binds Cdc42 to mediate transformation. Nature 2000, 405, 800–804. [Google Scholar] [CrossRef]
- Raghu, H.; Sharma-Walia, N.; Veettil, M.V.; Sadagopan, S.; Caballero, A.; Sivakumar, R.; Varga, L.; Bottero, V.; Chandran, B. Lipid rafts of primary endothelial cells are essential for Kaposi’s sarcoma-associated herpesvirus/human herpesvirus 8-induced phosphatidylinositol 3-kinase and RhoA-GTPases critical for microtubule dynamics and nuclear delivery of viral DNA but dispensable for binding and entry. J. Virol. 2007, 81, 7941–7959. [Google Scholar] [CrossRef] [Green Version]
- Döhner, K.; Nagel, C.-H.; Sodeik, B. Viral stop-and-go along microtubules: Taking a ride with dynein and kinesins. Trends Microbiol. 2005, 13, 320–327. [Google Scholar] [CrossRef] [PubMed]
- Alper, J.D.; Decker, F.; Agana, B.; Howard, J. The Motility of Axonemal Dynein Is Regulated by the Tubulin Code. Biophys. J. 2014, 107, 2872–2880. [Google Scholar] [CrossRef] [Green Version]
- Husain, M.; Harrod, K.S. Enhanced acetylation of alpha-tubulin in influenza A virus infected epithelial cells. FEBS Lett. 2011, 585, 128–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elliott, G.; O’Hare, P. Herpes Simplex Virus Type 1 Tegument Protein VP22 Induces the Stabilization and Hyperacetylation of Microtubules. J. Virol. 1998, 72, 6448–6455. [Google Scholar] [CrossRef] [Green Version]
- Atanasiu, D.; Saw, W.T.; Cohen, G.H.; Eisenberg, R.J. Cascade of Events Governing Cell-Cell Fusion Induced by Herpes Simplex Virus Glycoproteins gD, gH/gL, and gB. J. Virol. 2010, 84, 12292–12299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atanasiu, D.; Saw, W.T.; Eisenberg, R.J.; Cohen, G.H. Regulation of Herpes Simplex Virus Glycoprotein-Induced Cascade of Events Governing Cell-Cell Fusion. J. Virol. 2016, 90, 10535–10544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palazzo, A.; Ackerman, B.; Gundersen, G.G. Cell biology: Tubulin acetylation and cell motility. Nature 2003, 421, 230. [Google Scholar] [CrossRef]
- Naghavi, M.H.; Walsh, D. Microtubule Regulation and Function during Virus Infection. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [Green Version]
- Baghi, H.B.; Laval, K.; Favoreel, H.; Nauwynck, H.J. Isolation and characterization of equine nasal mucosal CD172a+ cells. Vet. Immunol. Immunopathol. 2014, 157, 155–163. [Google Scholar] [CrossRef]
- Nakamichi, K.; Matsumoto, Y.; Otsuka, H. Bovine Herpesvirus 1 Glycoprotein G Is Necessary for Maintaining Cell-to-Cell Junctional Adherence among Infected Cells. Virology 2002, 294, 22–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haller, C.; Tibroni, N.; Rudolph, J.M.; Grosse, R.; Fackler, O.T. Nef does not inhibit F-actin remodelling and HIV-1 cell–cell transmission at the T lymphocyte virological synapse. Eur. J. Cell Biol. 2011, 90, 913–921. [Google Scholar] [CrossRef] [PubMed]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kolyvushko, O.; Kelch, M.A.; Osterrieder, N.; Azab, W. Equine Alphaherpesviruses Require Activation of the Small GTPases Rac1 and Cdc42 for Intracellular Transport. Microorganisms 2020, 8, 1013. https://doi.org/10.3390/microorganisms8071013
Kolyvushko O, Kelch MA, Osterrieder N, Azab W. Equine Alphaherpesviruses Require Activation of the Small GTPases Rac1 and Cdc42 for Intracellular Transport. Microorganisms. 2020; 8(7):1013. https://doi.org/10.3390/microorganisms8071013
Chicago/Turabian StyleKolyvushko, Oleksandr, Maximilian A. Kelch, Nikolaus Osterrieder, and Walid Azab. 2020. "Equine Alphaherpesviruses Require Activation of the Small GTPases Rac1 and Cdc42 for Intracellular Transport" Microorganisms 8, no. 7: 1013. https://doi.org/10.3390/microorganisms8071013