Key Bacteria in the Gut Microbiota Network for the Transition between Sedentary and Active Lifestyle


 ,
,  ,
, 
{kind=link}
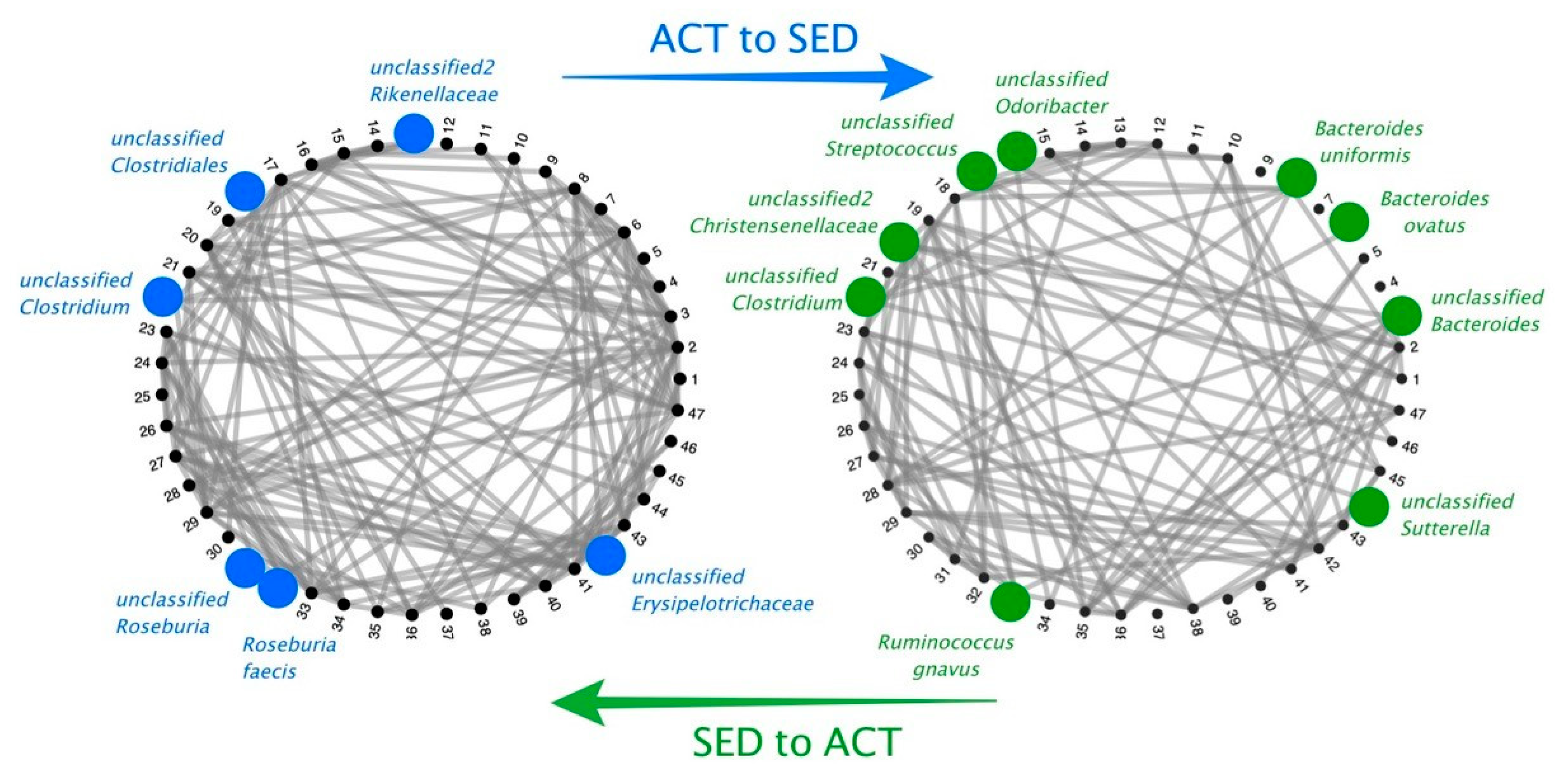{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Data Collection
2.2. Microbiota and Network Analysis
2.3. Statistical Analysis
3. Results
3.1. Population Characteristics
3.2. Network Topology
3.3. Conversion Networks
4. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- Fiuza-Luces, C.; Santos-Lozano, A.; Joyner, M.; Carrera-Bastos, P.; Picazo, O.; Zugaza, J.L.; Izquierdo, M.; Ruilope, L.M.; Lucia, A. Exercise benefits in cardiovascular disease: Beyond attenuation of traditional risk factors. Nat. Rev. Cardiol. 2018, 15, 731–743. [Google Scholar] [CrossRef] [PubMed]
- Thomas, R.J.; Kenfield, S.A.; Jimenez, A. Exercise-induced biochemical changes and their potential influence on cancer: A scientific review. Br. J. Sports Med. 2016, 51, 640–644. [Google Scholar] [CrossRef] [PubMed]
- González-Gross, M.; Meléndez, A. Sedentarism, active lifestyle and sport: Impact on health and obesity prevention. Nutr. Hosp. 2013, 28, 89–98. [Google Scholar]
- Ekelund, U.; Steene-Johannessen, J.; Brown, W.; Fagerland, M.W.; Owen, N.; Powell, K.E.; Bauman, A.; Lee, I.-M. Does physical activity attenuate, or even eliminate, the detrimental association of sitting time with mortality? A harmonised meta-analysis of data from more than 1 million men and women. Lancet 2016, 388, 1302–1310. [Google Scholar] [CrossRef] [Green Version]
- Hallal, P.R.C.; Andersen, L.B.; Bull, F.C.; Guthold, R.; Haskell, W.; Ekelund, U. Global physical activity levels: Surveillance progress, pitfalls, and prospects. Lancet 2012, 380, 247–257. [Google Scholar] [CrossRef]
- Cerdá, B.; Pérez, M.; Pérez-Santiago, J.D.; Tornero-Aguilera, J.F.; Gonzalez-Soltero, R.; Larrosa, M. Gut Microbiota Modification: Another Piece in the Puzzle of the Benefits of Physical Exercise in Health? Front. Physiol. 2016, 7, 42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mach, N.; Fuster-Botella, D. Endurance exercise and gut microbiota: A review. J. Sport Health Sci. 2016, 6, 179–197. [Google Scholar] [CrossRef]
- Castellanos, N.; Diez, G.G.; Antúnez-Almagro, C.; Bailén, M.; Bressa, C.; Soltero, R.G.; Pérez, M.; Larrosa, M. A Critical Mutualism—Competition Interplay Underlies the Loss of Microbial Diversity in Sedentary Lifestyle. Front. Microbiol. 2020, 10, 3142. [Google Scholar] [CrossRef]
- World Health Organization. Global Recommendations on Physical Activity for Health; WHO Press: Geneva, Switzerland, 2010. [Google Scholar]
- Quantitative Insights into Microbial Ecology (QIIME) Program, Version 1.9.1. Available online: http://qiime.org (accessed on 1 April 2020).
- Faust, K.; Raes, J. Microbial interactions: From networks to models. Nat. Rev. Genet. 2012, 10, 538–550. [Google Scholar] [CrossRef]
- Layeghifard, M.; Hwang, D.M.; Guttman, D.S. Disentangling Interactions in the Microbiome: A Network Perspective. Trends Microbiol. 2016, 25, 217–228. [Google Scholar] [CrossRef]
- Layeghifard, M.; Li, H.; Wang, P.W.; Donaldson, S.L.; Coburn, B.; Clark, S.T.; Caballero, J.D.; Zhang, Y.; Tullis, D.E.; Yau, Y.C.W.; et al. Microbiome networks and change-point analysis reveal key community changes associated with cystic fibrosis pulmonary exacerbations. NPJ Biofilms Microbiomes 2019, 5, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamanai-Shacoori, Z.; Smida, I.; Bousarghin, L.; Loreal, O.; Meuric, V.; Fong, S.B.; Bonnaure-Mallet, M.; Jolivet-Gougeon, A. Roseburia spp.: A marker of health? Future Microbiol. 2017, 12, 157–170. [Google Scholar] [CrossRef] [PubMed]
- La Rosa, S.L.; Leth, M.; Michalak, L.; Hansen, M.E.; Pudlo, N.A.; Glowacki, R.; Pereira, G.; Workman, C.T.; Arntzen, M.; Pope, P.B.; et al. The human gut Firmicute Roseburia intestinalis is a primary degrader of dietary β-mannans. Nat. Commun. 2019, 10, 905. [Google Scholar] [CrossRef] [PubMed]
- Estaki, M.; Pither, J.; Baumeister, P.; Little, J.P.; Gill, S.K.; Ghosh, S.; Ahmadi-Vand, Z.; Marsden, K.R.; Gibson, D.L. Cardiorespiratory fitness as a predictor of intestinal microbial diversity and distinct metagenomic functions. Microbiome 2016, 4, 42. [Google Scholar] [CrossRef] [Green Version]
- Bressa, C.; Bailen, M.; Pérez-Santiago, J.; Gonzalez-Soltero, R.; Pérez, M.; Montalvo-Lominchar, M.G.; Maté-Muñoz, J.L.; Domínguez, R.; Moreno, D.; Larrosa, M. Differences in gut microbiota profile between women with active lifestyle and sedentary women. PLoS ONE 2017, 12, e0171352. [Google Scholar] [CrossRef] [Green Version]
- Parthasarathy, G.; Chen, J.; Chen, X.; Chia, N.; O’Connor, H.M.; Wolf, P.G.; Gaskins, H.R.; Bharucha, A.E. Faculty Opinions recommendation of Relationship between microbiota of the colonic mucosa vs feces and symptoms, colonic transit, and methane production in female patients with chronic constipation. Gastroenterology 2016, 150, 367. [Google Scholar] [CrossRef] [Green Version]
- Vandeputte, D.; Falony, G.; Vieira-Silva, S.; Tito, R.Y.; Joossens, M.; Raes, J. Stool consistency is strongly associated with gut microbiota richness and composition, enterotypes and bacterial growth rates. Gut 2015, 65, 57–62. [Google Scholar] [CrossRef] [Green Version]
- Machiels, K.; Joossens, M.; Sabino, J.; De Preter, V.; Arijs, I.; Eeckhaut, V.; Ballet, V.; Claes, K.; Van Immerseel, F.; Verbeke, K.; et al. Faculty Opinions recommendation of A decrease of the butyrate-producing species Roseburia hominis and Faecalibacterium prausnitzii defines dysbiosis in patients with ulcerative colitis. Gut 2015, 63, 1275–1283. [Google Scholar] [CrossRef]
- Tilg, H.; Moschen, A.R. Microbiota and diabetes: An evolving relationship. Gut 2014, 63, 1513–1521. [Google Scholar] [CrossRef]
- Zhang, H.; DiBaise, J.K.; Zuccolo, A.; Kudrna, D.; Braidotti, M.; Yu, Y.; Parameswaran, P.; Crowell, M.; Wing, R.A.; Rittmann, B.E.; et al. Human gut microbiota in obesity and after gastric bypass. Proc. Natl. Acad. Sci. USA 2009, 106, 2365–2370. [Google Scholar] [CrossRef] [Green Version]
- Clarke, S.; Murphy, E.F.; O’Sullivan, O.; Lucey, A.; Humphreys, M.; Hogan, A.; Hayes, P.; O’Reilly, M.; Jeffery, I.B.; Wood-Martin, R.; et al. Exercise and associated dietary extremes impact on gut microbial diversity. Gut 2014, 63, 1913–1920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamoureux, E.V.; Grandy, S.; Langille, M.G.I. Moderate Exercise Has Limited but Distinguishable Effects on the Mouse Microbiome. mSystems 2017, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jangi, S.; Gandhi, R.; Cox, L.M.; Li, N.; Von Glehn, F.; Yan, R.; Patel, B.; Mazzola, M.A.; Liu, S.; Glanz, B.L.; et al. Alterations of the human gut microbiome in multiple sclerosis. Nat. Commun. 2016, 7, 12015. [Google Scholar] [CrossRef] [PubMed]
- Ding, H.T.; Taur, Y.; Walkup, J.T. Gut Microbiota and Autism: Key Concepts and Findings. J. Autism Dev. Disord. 2016, 47, 480–489. [Google Scholar] [CrossRef] [PubMed]
- Kaakoush, N.O. Sutterella Species, IgA-degrading Bacteria in Ulcerative Colitis. Trends Microbiol. 2020. [Google Scholar] [CrossRef]
- Gaike, A.H.; Paul, D.; Bhute, S.; Dhotre, D.P.; Pande, P.; Upadhyaya, S.; Reddy, Y.; Sampath, R.; Ghosh, D.; Chandraprabha, D.; et al. The Gut Microbial Diversity of Newly Diagnosed Diabetics but Not of Prediabetics Is Significantly Different from That of Healthy Nondiabetics. mSystems 2020, 5. [Google Scholar] [CrossRef] [Green Version]
- Henke, M.T.; Kenny, D.; Cassilly, C.D.; Vlamakis, H.; Xavier, R.J.; Clardy, J. Ruminococcus gnavus, a member of the human gut microbiome associated with Crohn’s disease, produces an inflammatory polysaccharide. Proc. Natl. Acad. Sci. USA 2019, 116, 12672–12677. [Google Scholar] [CrossRef] [Green Version]
- Hall, A.B.; Yassour, M.; Sauk, J.; Garner, A.; Jiang, X.; Arthur, T.; Lagoudas, G.K.; Vatanen, T.; Fornelos, N.; Wilson, R.; et al. A novel Ruminococcus gnavus clade enriched in inflammatory bowel disease patients. Genome Med. 2017, 9, 103. [Google Scholar] [CrossRef]
- Clark, A.; Mach, N. Exercise-induced stress behavior, gut-microbiota-brain axis and diet: A systematic review for athletes. J. Int. Soc. Sports Nutr. 2016, 13, 43. [Google Scholar] [CrossRef] [Green Version]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Castellanos, N.; Diez, G.G.; Antúnez-Almagro, C.; Bressa, C.; Bailén, M.; González-Soltero, R.; Pérez, M.; Larrosa, M. Key Bacteria in the Gut Microbiota Network for the Transition between Sedentary and Active Lifestyle. Microorganisms 2020, 8, 785. https://doi.org/10.3390/microorganisms8050785
Castellanos N, Diez GG, Antúnez-Almagro C, Bressa C, Bailén M, González-Soltero R, Pérez M, Larrosa M. Key Bacteria in the Gut Microbiota Network for the Transition between Sedentary and Active Lifestyle. Microorganisms. 2020; 8(5):785. https://doi.org/10.3390/microorganisms8050785
Chicago/Turabian StyleCastellanos, Nazareth, Gustavo G. Diez, Carmen Antúnez-Almagro, Carlo Bressa, María Bailén, Rocío González-Soltero, Margarita Pérez, and Mar Larrosa. 2020. "Key Bacteria in the Gut Microbiota Network for the Transition between Sedentary and Active Lifestyle" Microorganisms 8, no. 5: 785. https://doi.org/10.3390/microorganisms8050785