Insight into the Lifestyle of Amoeba Willaertia magna during Bioreactor Growth Using Transcriptomics and Proteomics

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. W. magna C2c maky Culture in Bioreactor
2.2. Illumina Sequencing of the Transcriptome
2.3. Transcriptomic Analysis of W. Magna C2c Maky
2.4. Functional Annotation
2.5. Proteomics
2.5.1. Cell Lysis
2.5.2. SDS-Page and Western-Blot
2.5.3. Liquid Digestion
2.5.4. NanoLC-MS/MS Analysis
2.6. Data Analysis
3. Results
3.1. Transcriptomic Analysis
3.2. Mean Proteomic Information
3.3. COG Enrichment Analysis
3.4. KEGG Enrichment Analysis
3.5. W. magna Shape and Movement
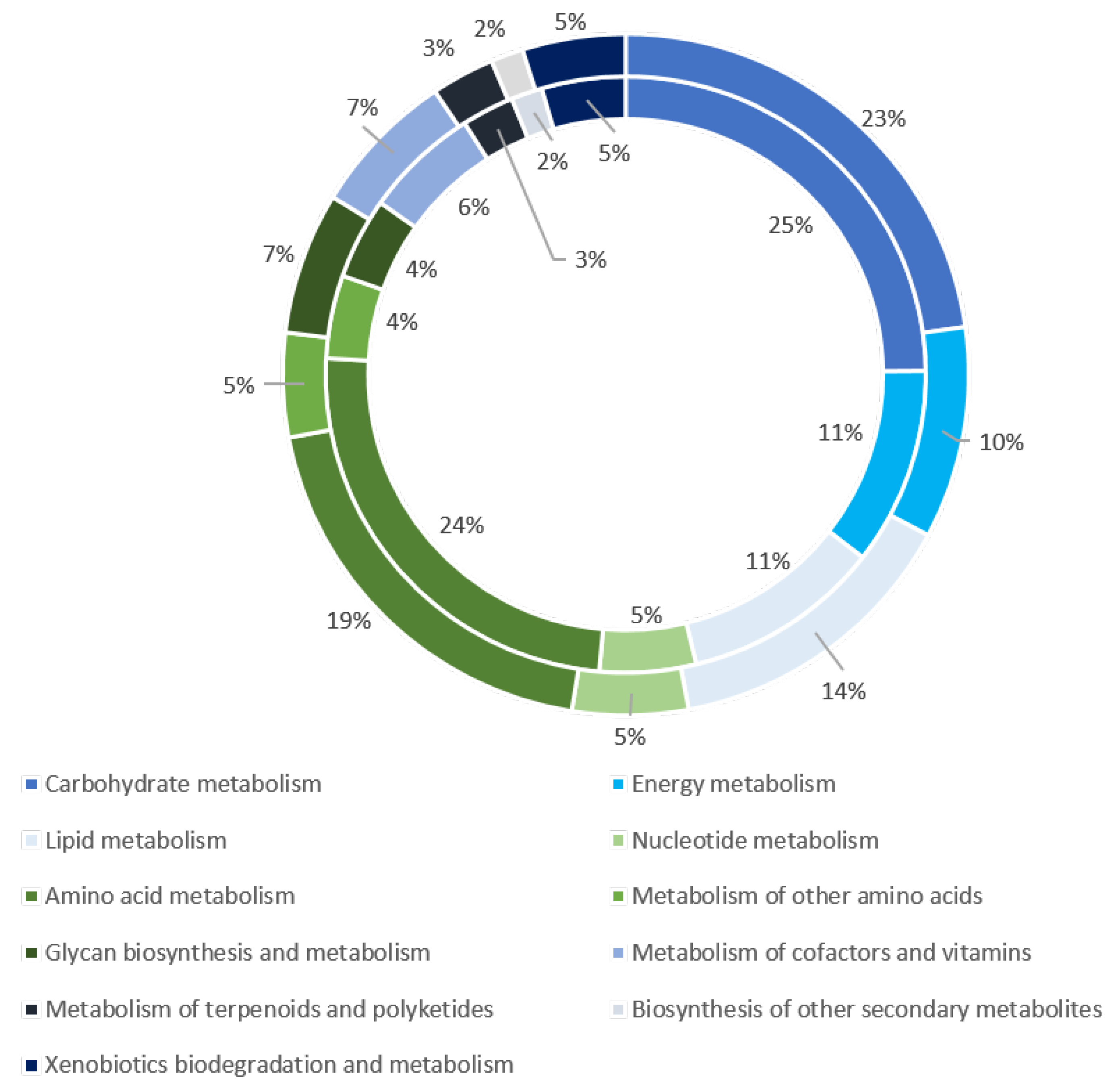
3.6. Metabolism
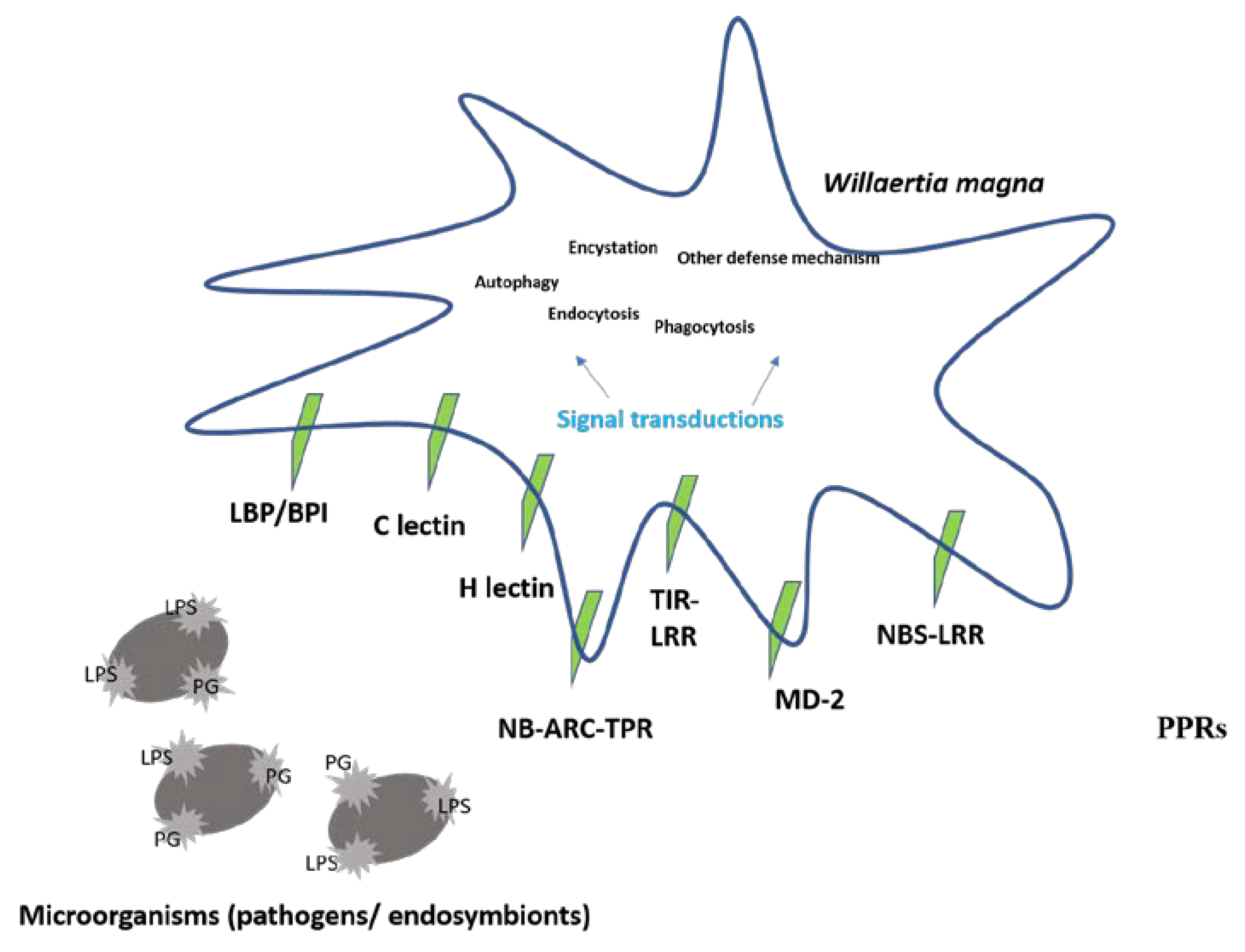
3.7. Defense Mechanisms
3.8. Ecological Context
4. Discussion
5. Conclusions
Supplementary Materials
Availability of Data
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Adl, S.M.; Bass, D.; Lane, C.E.; Lukeš, J.; Schoch, C.L.; Smirnov, A.; Agatha, S.; Berney, C.; Brown, M.W.; Burki, F.; et al. Revisions to the Classification, Nomenclature, and Diversity of Eukaryotes. J. Eukaryot. Microbiol. 2018, jeu.12691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertelli, C.; Greub, G. Lateral gene exchanges shape the genomes of amoeba-resisting microorganisms. Front. Cell. Infect. Microbiol. 2012, 2. [Google Scholar] [CrossRef] [Green Version]
- Pánek, T.; Čepička, I. Diversity of Heterolobosea. Genet. Divers. Microorg. 2012. [Google Scholar] [CrossRef] [Green Version]
- de Jonckheere, J.F.; Dive, D.G.; Pussard, M.; Vickerman, K. Willaertia Magna gen. nov., sp. nov. (Vahlkampfiidae), a Thermophilic Amoeba Found in Different Habitats. Available online: https://eurekamag.com/research/001/281/001281223.php (accessed on 12 August 2018).
- Robinson, B.S.; Christy, P.E.; De Jonckheere, J.F. A temporary flagellate (mastigote) stage in the vahlkampfiid amoeba Willaertia magna and its possible evolutionary significance. Biosystems 1989, 23, 75–86. [Google Scholar] [CrossRef]
- Clarholm, M. Protozoan grazing of bacteria in soil—impact and importance. Microb. Ecol. 1981, 7, 343–350. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Zaragoza, S. Ecology of Free-Living Amoebae. Crit. Rev. Microbiol. 1994, 20, 225–241. [Google Scholar] [CrossRef]
- Greub, G.; Raoult, D. Microorganisms Resistant to Free-Living Amoebae. Clin. Microbiol. Rev. 2004, 17, 413–433. [Google Scholar] [CrossRef] [Green Version]
- Rowbotham, T.J. Preliminary report on the pathogenicity of Legionella pneumophila for freshwater and soil amoebae. J. Clin. Pathol. 1980, 33, 1179–1183. [Google Scholar] [CrossRef] [Green Version]
- Rowbotham, T.J. Isolation of Legionella pneumophila from clinical specimens via amoebae, and the interaction of those and other isolates with amoebae. J. Clin. Pathol. 1983, 36, 978–986. [Google Scholar] [CrossRef] [Green Version]
- Stout, J.E.; Yu, V.L. Legionellosis. N. Engl. J. Med. 1997, 337, 682–687. [Google Scholar] [CrossRef]
- Barbaree, J.M.; Fields, B.S.; Feeley, J.C.; Gorman, G.W.; Martin, W.T. Isolation of protozoa from water associated with a legionellosis outbreak and demonstration of intracellular multiplication of Legionella pneumophila. Appl. Environ. Microbiol. 1986, 51, 422–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scheikl, U.; Sommer, R.; Kirschner, A.; Rameder, A.; Schrammel, B.; Zweimüller, I.; Wesner, W.; Hinker, M.; Walochnik, J. Free-living amoebae (FLA) co-occurring with legionellae in industrial waters. Eur. J. Protistol. 2014, 50, 422–429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dupuy, M.; Binet, M.; Bouteleux, C.; Herbelin, P.; Soreau, S.; Héchard, Y. Permissiveness of freshly isolated environmental strains of amoebae for growth of Legionella pneumophila. FEMS Microbiol. Lett. 2016, 363, fnw022. [Google Scholar] [CrossRef] [PubMed]
- Kilvington, S.; Stevens, C.; Ebert, F.; Michel, R.; Beeching, J.R. A comparative study of Willaertia magna (free-living amoeba) from different geographic areas using whole-cell and small-subunit rRNA restriction fragment length polymorphisms. J. Protozool. Res. 1995, 5, 97–107. [Google Scholar]
- Linder, J.W.-K. Ewert Free-living Amoebae Protecting Legionella in Water: The Tip of an Iceberg? Scand. J. Infect. Dis. 1999, 31, 383–385. [Google Scholar] [CrossRef]
- Cirillo, J.D.; Falkow, S.; Tompkins, L.S. Growth of Legionella pneumophila in Acanthamoeba castellanii enhances invasion. Infect. Immun. 1994, 62, 3254–3261. [Google Scholar] [CrossRef] [Green Version]
- Delafont, V.; Rodier, M.-H.; Maisonneuve, E.; Cateau, E. Vermamoeba vermiformis: A Free-Living Amoeba of Interest. Microb. Ecol. 2018, 76, 991–1001. [Google Scholar] [CrossRef]
- Dey, R.; Bodennec, J.; Mameri, M.O.; Pernin, P. Free-living freshwater amoebae differ in their susceptibility to the pathogenic bacterium Legionella pneumophila. FEMS Microbiol. Lett. 2009, 290, 10–17. [Google Scholar] [CrossRef] [Green Version]
- Hasni, I.; Jarry, A.; Quelard, B.; Carlino, A.; Eberst, J.-B.; Abbe, O.; Demanèche, S. Intracellular Behaviour of Three Legionella pneumophila Strains within Three Amoeba Strains, Including Willaertia magna C2c Maky. Pathogens 2020, 9, 105. [Google Scholar] [CrossRef] [Green Version]
- Amoeba|Biocide by Nature. Available online: http://www.amoeba-biocide.com/fr (accessed on 7 January 2020).
- Neff, R.J. Purification, Axenic Cultivation, and Description of a Soil Amoeba, Acanthamoeba sp. J. Protozool. 1957, 4, 176–182. [Google Scholar] [CrossRef]
- Weekers, P.H.H.; Vogels, G.D. Axenic cultivation of the free-living amoebae, Acanthamoeba castellanii and Hartmannella vermiformis in a chemostat. J. Microbiol. Methods 1994, 19, 13–18. [Google Scholar] [CrossRef]
- Beshay, U.; Friehs, K.; Azzam, A.-E.-M.; Flaschel, E. Analysis of the behaviour of Dictyostelium discoideum in immobilised state by means of continuous cultivation. Bioprocess. Biosyst. Eng. 2003, 26, 117–122. [Google Scholar] [CrossRef] [PubMed]
- Mimouni, V.; Ulmann, L.; Pasquet, V.; Mathieu, M.; Picot, L.; Bougaran, G.; Cadoret, J.-P.; Morant-Manceau, A.; Schoefs, B. The potential of microalgae for the production of bioactive molecules of pharmaceutical interest. Curr. Pharm. Biotechnol. 2012, 13, 2733–2750. [Google Scholar] [CrossRef] [PubMed]
- Newman, J.D.; Marshall, J.; Chang, M.; Nowroozi, F.; Paradise, E.; Pitera, D.; Newman, K.L.; Keasling, J.D. High-level production of amorpha-4,11-diene in a two-phase partitioning bioreactor of metabolically engineered Escherichia coli. Biotechnol. Bioeng. 2006, 95, 684–691. [Google Scholar] [CrossRef] [PubMed]
- Papaspyridi, L.-M.; Aligiannis, N.; Christakopoulos, P.; Skaltsounis, A.-L.; Fokialakis, N. Production of bioactive metabolites with pharmaceutical and nutraceutical interest by submerged fermentation of Pleurotus ostreatus in a batch stirred tank bioreactor. Procedia Food Sci. 2011, 1, 1746–1752. [Google Scholar] [CrossRef] [Green Version]
- Hasni, I.; Chelkha, N.; Baptiste, E.; Mameri, M.R.; Lachuer, J.; Plasson, F.; Colson, P.; Scola, B.L. Investigation of potential pathogenicity of Willaertia magna by investigating the transfer of bacteria pathogenicity genes into its genome. Sci. Rep. 2019, 9, 1–12. [Google Scholar] [CrossRef]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef]
- Lomsadze, A.; Burns, P.D.; Borodovsky, M. Integration of mapped RNA-Seq reads into automatic training of eukaryotic gene finding algorithm. Nucleic Acids Res. 2014, 42, e119. [Google Scholar] [CrossRef]
- Vogel, C.; Marcotte, E.M. Insights into the regulation of protein abundance from proteomic and transcriptomic analyses. Nat. Rev. Genet. 2012, 13, 227–232. [Google Scholar] [CrossRef]
- Fritz-Laylin, L.K.; Prochnik, S.E.; Ginger, M.L.; Dacks, J.B.; Carpenter, M.L.; Field, M.C.; Kuo, A.; Paredez, A.; Chapman, J.; Pham, J.; et al. The Genome of Naegleria gruberi Illuminates Early Eukaryotic Versatility. Cell 2010, 140, 631–642. [Google Scholar] [CrossRef] [Green Version]
- Opperdoes, F.R.; De Jonckheere, J.F.; Tielens, A.G.M. Naegleria gruberi metabolism. Int. J. Parasitol. 2011, 41, 915–924. [Google Scholar] [CrossRef] [PubMed]
- Alsam, S.; Sissons, J.; Dudley, R.; Khan, N.A. Mechanisms associated with Acanthamoeba castellanii (T4) phagocytosis. Parasitol. Res. 2005, 96, 402–409. [Google Scholar] [CrossRef]
- Clarke, M.; Lohan, A.J.; Liu, B.; Lagkouvardos, I.; Roy, S.; Zafar, N.; Bertelli, C.; Schilde, C.; Kianianmomeni, A.; Bürglin, T.R.; et al. Genome of Acanthamoeba castellanii highlights extensive lateral gene transfer and early evolution of tyrosine kinase signaling. Genome Biol. 2013, 14, R11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, N.A. Acanthamoeba: Biology and increasing importance in human health. FEMS Microbiol. Rev. 2006, 30, 564–595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Jonckheere, J. Use of an axenic medium for differentiation between pathogenic and nonpathogenic Naegleria fowleri isolates. Appl. Environ. Microbiol. 1977, 33, 751–757. [Google Scholar] [CrossRef] [Green Version]
- Bioréacteur Pilote GPC. Available online: http://www.gpcbio.com/bioracteurpilote.html (accessed on 21 March 2020).
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Aherfi, S.; Andreani, J.; Baptiste, E.; Oumessoum, A.; Dornas, F.P.; Andrade, A.C.; Chabriere, E.; Abrahao, J.; Levasseur, A.; Raoult, D.; et al. A Large Open Pangenome and a Small Core Genome for Giant Pandoraviruses. Front. Microbiol. 2018, 9. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [Green Version]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—a Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef]
- Huerta-Cepas, J.; Forslund, K.; Coelho, L.P.; Szklarczyk, D.; Jensen, L.J.; von Mering, C.; Bork, P. Fast Genome-Wide Functional Annotation through Orthology Assignment by eggNOG-Mapper. Mol. Biol. Evol. 2017, 34, 2115–2122. [Google Scholar] [CrossRef] [Green Version]
- Tatusov, R.L.; Galperin, M.Y.; Natale, D.A.; Koonin, E.V. The COG database: A tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res. 2000, 28, 33–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanehisa, M.; Goto, S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Marchler-Bauer, A.; Derbyshire, M.K.; Gonzales, N.R.; Lu, S.; Chitsaz, F.; Geer, L.Y.; Geer, R.C.; He, J.; Gwadz, M.; Hurwitz, D.I.; et al. CDD: NCBI’s conserved domain database. Nucleic Acids Res. 2015, 43, D222–D226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hunter, S.; Apweiler, R.; Attwood, T.K.; Bairoch, A.; Bateman, A.; Binns, D.; Bork, P.; Das, U.; Daugherty, L.; Duquenne, L.; et al. InterPro: The integrative protein signature database. Nucleic Acids Res. 2009, 37, D211–D215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bachmann, B.O.; Ravel, J. Chapter 8. Methods for in silico prediction of microbial polyketide and nonribosomal peptide biosynthetic pathways from DNA sequence data. Methods Enzymol. 2009, 458, 181–217. [Google Scholar] [CrossRef]
- Ziemert, N.; Podell, S.; Penn, K.; Badger, J.H.; Allen, E.; Jensen, P.R. The natural product domain seeker NaPDoS: A phylogeny based bioinformatic tool to classify secondary metabolite gene diversity. PLoS ONE 2012, 7, e34064. [Google Scholar] [CrossRef] [Green Version]
- Almagro Armenteros, J.J.; Salvatore, M.; Emanuelsson, O.; Winther, O.; von Heijne, G.; Elofsson, A.; Nielsen, H. Detecting sequence signals in targeting peptides using deep learning. Life Sci. Alliance 2019, 2. [Google Scholar] [CrossRef] [Green Version]
- Prokisch, H.; Andreoli, C.; Ahting, U.; Heiss, K.; Ruepp, A.; Scharfe, C.; Meitinger, T. MitoP2: The mitochondrial proteome database—now including mouse data. Nucleic Acids Res. 2006, 34, D705–D711. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [Green Version]
- Letunic, I.; Bork, P. Interactive tree of life (iTOL) v3: An online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res. 2016, 44, W242–W245. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Diedrich, J.; Chu, Y.-Y.; Yates, J.R. Extracting Accurate Precursor Information for Tandem Mass Spectra by RawConverter. Anal. Chem. 2015, 87, 11361–11367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, B.; Zhang, K.; Hendrie, C.; Liang, C.; Li, M.; Doherty-Kirby, A.; Lajoie, G. PEAKS: Powerful software for peptide de novo sequencing by tandem mass spectrometry. Rapid Commun. Mass Spectrom. RCM 2003, 17, 2337–2342. [Google Scholar] [CrossRef] [PubMed]
- McCafferty, J.; Reid, R.; Spencer, M.; Hamp, T.; Fodor, A. Peak Studio: A tool for the visualization and analysis of fragment analysis files. Environ. Microbiol. Rep. 2012, 4, 556–561. [Google Scholar] [CrossRef] [PubMed]
- Khan, N.A. Acanthamoeba: Biology and Pathogenesis, 2nd ed.; Caister Academic Press: Norfolk, UK, 2018. [Google Scholar]
- Otomo, T.; Otomo, C.; Tomchick, D.R.; Machius, M.; Rosen, M.K. Structural Basis of Rho GTPase-Mediated Activation of the Formin mDia1. Mol. Cell 2005, 18, 273–281. [Google Scholar] [CrossRef] [PubMed]
- Hug, L.A.; Stechmann, A.; Roger, A.J. Phylogenetic Distributions and Histories of Proteins Involved in Anaerobic Pyruvate Metabolism in Eukaryotes. Mol. Biol. Evol. 2010, 27, 311–324. [Google Scholar] [CrossRef]
- Elsbach, P. The bactericidal/permeability-increasing protein (BPI) in antibacterial host defense. J. Leukoc. Biol. 1998, 64, 14–18. [Google Scholar] [CrossRef]
- Cosson, P.; Lima, W.C. Intracellular killing of bacteria: Is D ictyostelium a model macrophage or an alien?: Intracellular bacterial killing in Dictyostelium. Cell. Microbiol. 2014, 16, 816–823. [Google Scholar] [CrossRef] [Green Version]
- Winterbourn, C.C.; Kettle, A.J. Redox Reactions and Microbial Killing in the Neutrophil Phagosome. Antioxid. Redox Signal. 2012, 18, 642–660. [Google Scholar] [CrossRef]
- Hong, Y.; Kang, J.-M.; Joo, S.-Y.; Song, S.-M.; Lê, H.G.; Thái, T.L.; Lee, J.; Goo, Y.-K.; Chung, D.-I.; Sohn, W.-M.; et al. Molecular and Biochemical Properties of a Cysteine Protease of Acanthamoeba castellanii. Korean J. Parasitol. 2018, 56, 409–418. [Google Scholar] [CrossRef]
- Turk, V.; Stoka, V.; Vasiljeva, O.; Renko, M.; Sun, T.; Turk, B.; Turk, D. Cysteine cathepsins: From structure, function and regulation to new frontiers. Biochim. Biophys. Acta 2012, 1824, 68–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyanaga, A. Structure and function of polyketide biosynthetic enzymes: Various strategies for production of structurally diverse polyketides. Biosci. Biotechnol. Biochem. 2017, 81, 2227–2236. [Google Scholar] [CrossRef] [Green Version]
- Robinson, J.A. Polyketide synthase complexes: Their structure and function in antibiotic biosynthesis. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 1991, 332, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Singh, D.R. BIODEGRADATION OF XENOBIOTICS- A WAY FOR ENVIRONMENTAL DETOXIFICATION. Int. J. Dev. Res. 2017, 7, 14082–14087. [Google Scholar]
- Miranda, E.R.; Nam, E.A.; Kuspa, A.; Shaulsky, G. The ABC transporter, AbcB3, mediates cAMP export in D. discoideum development. Dev. Biol. 2015, 397, 203–211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eichinger, L.; Pachebat, J.A.; Glöckner, G.; Rajandream, M.-A.; Sucgang, R.; Berriman, M.; Song, J.; Olsen, R.; Szafranski, K.; Xu, Q.; et al. The genome of the social amoeba Dictyostelium discoideum. Nature 2005, 435, 43–57. [Google Scholar] [CrossRef] [Green Version]
- Brown, S.; Clastre, M.; Courdavault, V.; O’Connor, S.E. De novo production of the plant-derived alkaloid strictosidine in yeast. Proc. Natl. Acad. Sci. USA 2015, 112, 3205–3210. [Google Scholar] [CrossRef] [Green Version]
- Zysset-Burri, D.C.; Müller, N.; Beuret, C.; Heller, M.; Schürch, N.; Gottstein, B.; Wittwer, M. Genome-wide identification of pathogenicity factors of the free-living amoeba Naegleria fowleri. BMC Genomics 2014, 15. [Google Scholar] [CrossRef] [Green Version]
- Liechti, N.; Schürch, N.; Bruggmann, R.; Wittwer, M. The genome of Naegleria lovaniensis, the basis for a comparative approach to unravel pathogenicity factors of the human pathogenic amoeba N. fowleri. BMC Genomics 2018, 19. [Google Scholar] [CrossRef] [Green Version]
- Hasni, I.; Andréani, J.; Colson, P.; La Scola, B. Description of Virulent Factors and Horizontal Gene Transfers of Keratitis-Associated Amoeba Acanthamoeba Triangularis by Genome Analysis. Pathogens 2020, 9, 217. [Google Scholar] [CrossRef] [Green Version]
- Chelkha, N.; Hasni, I.; Louazani, A.C.; Levasseur, A.; La Scola, B.; Colson, P. Vermamoeba vermiformis CDC-19 draft genome sequence reveals considerable gene trafficking including with candidate phyla radiation and giant viruses. Sci. Rep. 2020, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Detering, H.; Aebischer, T.; Dabrowski, P.W.; Radonić, A.; Nitsche, A.; Renard, B.Y.; Kiderlen, A.F. First Draft Genome Sequence of Balamuthia mandrillaris, the Causative Agent of Amoebic Encephalitis. Genome Announc. 2015, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greninger, A.L.; Messacar, K.; Dunnebacke, T.; Naccache, S.N.; Federman, S.; Bouquet, J.; Mirsky, D.; Nomura, Y.; Yagi, S.; Glaser, C.; et al. Clinical metagenomic identification of Balamuthia mandrillaris encephalitis and assembly of the draft genome: The continuing case for reference genome sequencing. Genome Med. 2015, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, M.; Sun, X.; Xu, N.; Liao, Z.; Li, Y.; Wang, J.; Fan, Y.; Cui, D.; Li, P.; Miao, Z. Integration of deep transcriptome and proteome analyses of salicylic acid regulation high temperature stress in Ulva prolifera. Sci. Rep. 2017, 7, 1–19. [Google Scholar] [CrossRef]
- Lee, M.V.; Topper, S.E.; Hubler, S.L.; Hose, J.; Wenger, C.D.; Coon, J.J.; Gasch, A.P. A dynamic model of proteome changes reveals new roles for transcript alteration in yeast. Mol. Syst. Biol. 2011, 7, 514. [Google Scholar] [CrossRef]
- de Sousa Abreu, R.; Penalva, L.O.; Marcotte, E.M.; Vogel, C. Global signatures of protein and mRNA expression levels. Mol. Biosyst. 2009, 5, 1512–1526. [Google Scholar] [CrossRef] [Green Version]
- Fletcher, D.A.; Mullins, R.D. Cell mechanics and the cytoskeleton. Nature 2010, 463, 485–492. [Google Scholar] [CrossRef] [Green Version]
- Tekle Yonas, I.; Williams Jessica, R. Cytoskeletal architecture and its evolutionary significance in amoeboid eukaryotes and their mode of locomotion. R. Soc. Open Sci. 2016, 3, 160283. [Google Scholar] [CrossRef] [Green Version]
- Atteia, A.; van Lis, R.; Gelius-Dietrich, G.; Adrait, A.; Garin, J.; Joyard, J.; Rolland, N.; Martin, W. Pyruvate formate-lyase and a novel route of eukaryotic ATP synthesis in Chlamydomonas mitochondria. J. Biol. Chem. 2006, 281, 9909–9918. [Google Scholar] [CrossRef] [Green Version]
- Barberà, M.J.; Ruiz-Trillo, I.; Tufts, J.Y.A.; Bery, A.; Silberman, J.D.; Roger, A.J. Sawyeria marylandensis (Heterolobosea) has a hydrogenosome with novel metabolic properties. Eukaryot. Cell 2010, 9, 1913–1924. [Google Scholar] [CrossRef] [Green Version]
- Tsaousis, A.D.; Nývltová, E.; Šuták, R.; Hrdý, I.; Tachezy, J. A Nonmitochondrial Hydrogen Production in Naegleria gruberi. Genome Biol. Evol. 2014, 6, 792–799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siddiqui, R.; Khan, N.A. War of the microbial worlds: Who is the beneficiary in Acanthamoeba–bacterial interactions? Exp. Parasitol. 2012, 130, 311–313. [Google Scholar] [CrossRef] [PubMed]
- Denet, E.; Coupat-Goutaland, B.; Nazaret, S.; Pélandakis, M.; Favre-Bonté, S. Diversity of free-living amoebae in soils and their associated human opportunistic bacteria. Parasitol. Res. 2017, 116, 3151–3162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akya, A.; Pointon, A.; Thomas, C. Mechanism involved in phagocytosis and killing of Listeria monocytogenes by Acanthamoeba polyphaga. Parasitol. Res. 2009, 105, 1375–1383. [Google Scholar] [CrossRef]
- Tishkov, V.I.; Savin, S.S.; Yasnaya, A.S. Protein Engineering of Penicillin Acylase. Acta Nat. 2010, 2, 47–61. [Google Scholar] [CrossRef] [Green Version]
- Iqbal, J.; Siddiqui, R.; Khan, N.A. Acanthamoeba and bacteria produce antimicrobials to target their counterpart. Parasit. Vectors 2014, 7, 56. [Google Scholar] [CrossRef] [Green Version]
- Zucko, J.; Skunca, N.; Curk, T.; Zupan, B.; Long, P.F.; Cullum, J.; Kessin, R.H.; Hranueli, D. Polyketide synthase genes and the natural products potential of Dictyostelium discoideum. Bioinforma. Oxf. Engl. 2007, 23, 2543–2549. [Google Scholar] [CrossRef] [Green Version]
- Rozgaj, R. [Microbial degradation of xenobiotics in the environment]. Arh. Hig. Rada Toksikol. 1994, 45, 189–198. [Google Scholar]
- Janssen, D.B.; Dinkla, I.J.T.; Poelarends, G.J.; Terpstra, P. Bacterial degradation of xenobiotic compounds: Evolution and distribution of novel enzyme activities. Environ. Microbiol. 2005, 7, 1868–1882. [Google Scholar] [CrossRef] [Green Version]
- Li, W.V.; Li, J.J. Modeling and analysis of RNA-seq data: A review from a statistical perspective. Quant. Biol. Beijing China 2018, 6, 195–209. [Google Scholar] [CrossRef]
- Manga, P.; Klingeman, D.M.; Lu, T.-Y.S.; Mehlhorn, T.L.; Pelletier, D.A.; Hauser, L.J.; Wilson, C.M.; Brown, S.D. Replicates, Read Numbers, and Other Important Experimental Design Considerations for Microbial RNA-seq Identified Using Bacillus thuringiensis Datasets. Front. Microbiol. 2016, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schurch, N.J.; Schofield, P.; Gierliński, M.; Cole, C.; Sherstnev, A.; Singh, V.; Wrobel, N.; Gharbi, K.; Simpson, G.G.; Owen-Hughes, T.; et al. How many biological replicates are needed in an RNA-seq experiment and which differential expression tool should you use? RNA 2016, 22, 839–851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rapaport, F.; Khanin, R.; Liang, Y.; Pirun, M.; Krek, A.; Zumbo, P.; Mason, C.E.; Socci, N.D.; Betel, D. Comprehensive evaluation of differential gene expression analysis methods for RNA-seq data. Genome Biol. 2013, 14, 3158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Zhou, J.; White, K.P. RNA-seq differential expression studies: More sequence or more replication? Bioinformatics 2014, 30, 301–304. [Google Scholar] [CrossRef]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hasni, I.; Decloquement, P.; Demanèche, S.; Mameri, R.M.; Abbe, O.; Colson, P.; La Scola, B. Insight into the Lifestyle of Amoeba Willaertia magna during Bioreactor Growth Using Transcriptomics and Proteomics. Microorganisms 2020, 8, 771. https://doi.org/10.3390/microorganisms8050771
Hasni I, Decloquement P, Demanèche S, Mameri RM, Abbe O, Colson P, La Scola B. Insight into the Lifestyle of Amoeba Willaertia magna during Bioreactor Growth Using Transcriptomics and Proteomics. Microorganisms. 2020; 8(5):771. https://doi.org/10.3390/microorganisms8050771
Chicago/Turabian StyleHasni, Issam, Philippe Decloquement, Sandrine Demanèche, Rayane Mouh Mameri, Olivier Abbe, Philippe Colson, and Bernard La Scola. 2020. "Insight into the Lifestyle of Amoeba Willaertia magna during Bioreactor Growth Using Transcriptomics and Proteomics" Microorganisms 8, no. 5: 771. https://doi.org/10.3390/microorganisms8050771