Molecular Ecological Network Complexity Drives Stand Resilience of Soil Bacteria to Mining Disturbances among Typical Damaged Ecosystems in China



Abstract
:1. Introduction
2. Materials and Methods
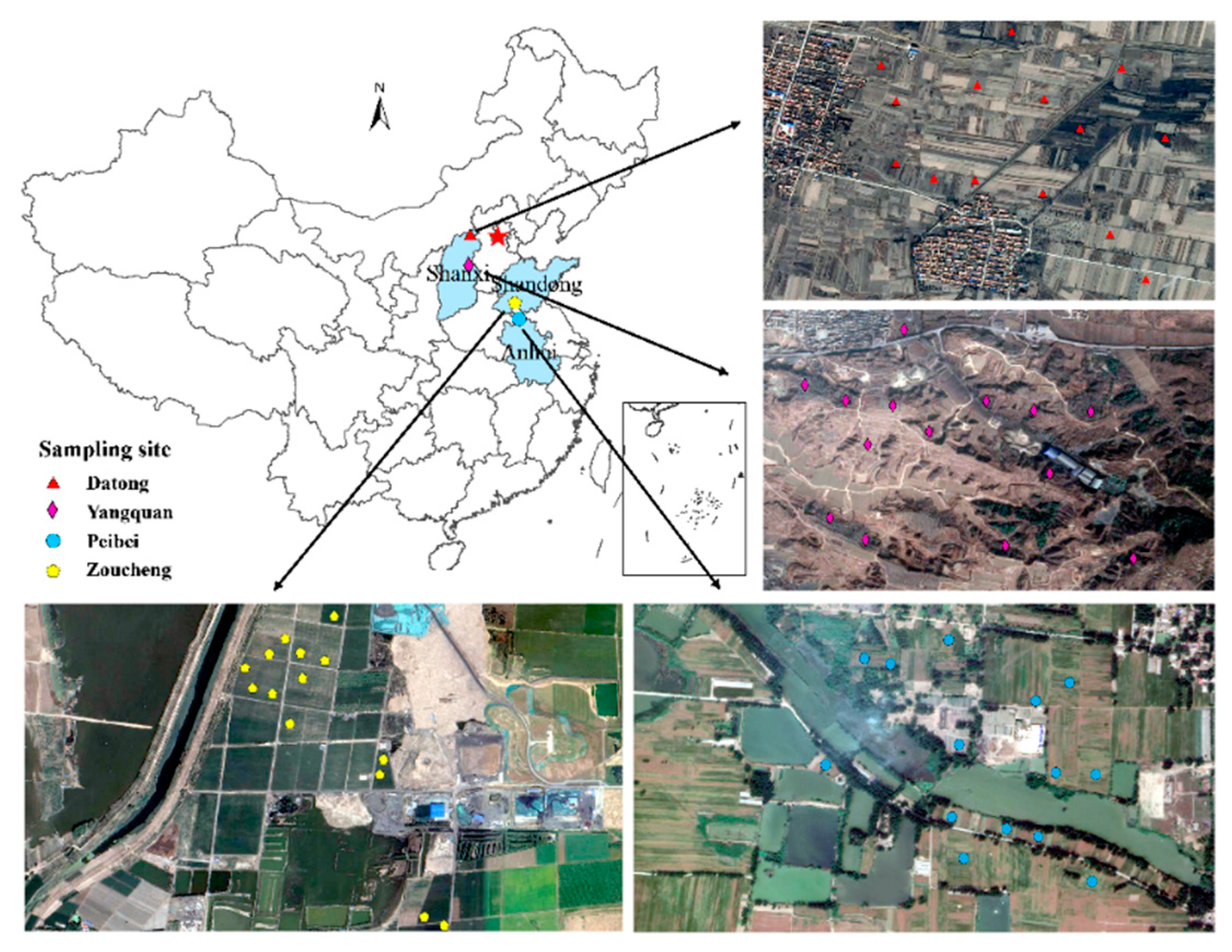
2.1. Study Sites, Soil Sampling, and Measurment
2.2. DNA Extraction, PCR Amplification, and Illumina MiSeq Sequencing
2.3. Network Construction and Analysis
2.4. Characterization of the Molecular Ecological Networks and Statistical Analysis
3. Results
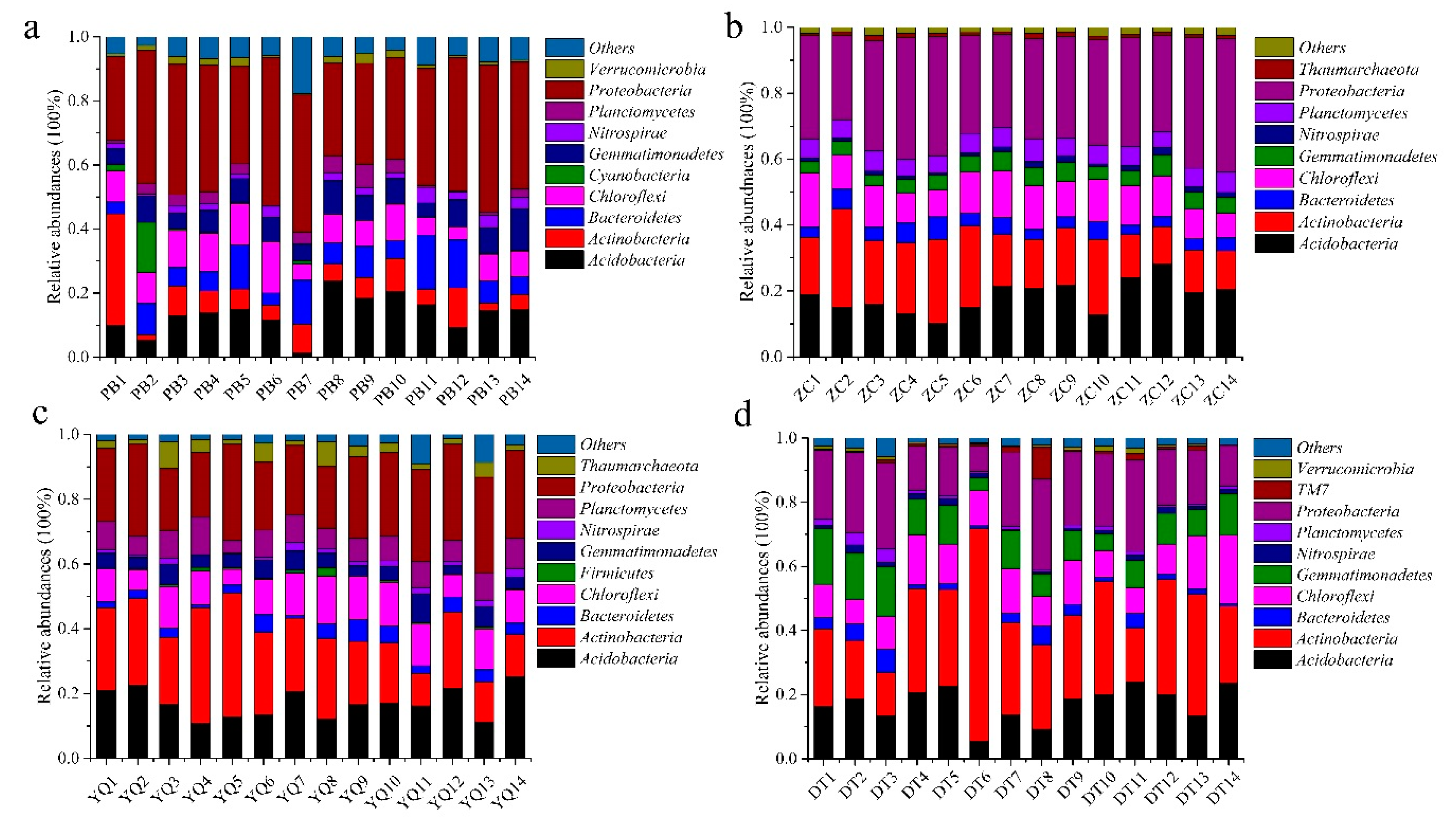
3.1. The Taxonomic Composition of Microbial Consortia in Different Mining Areas
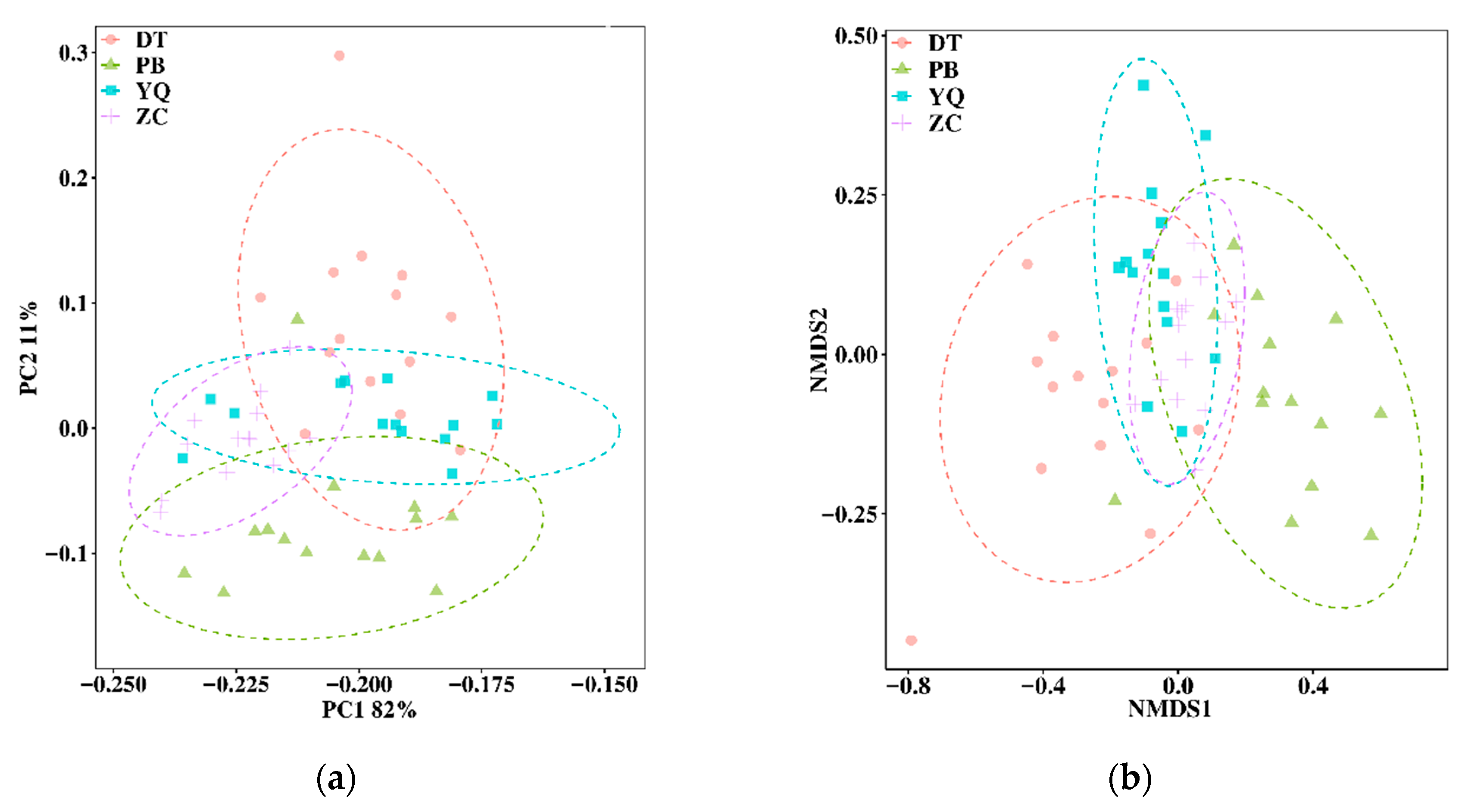
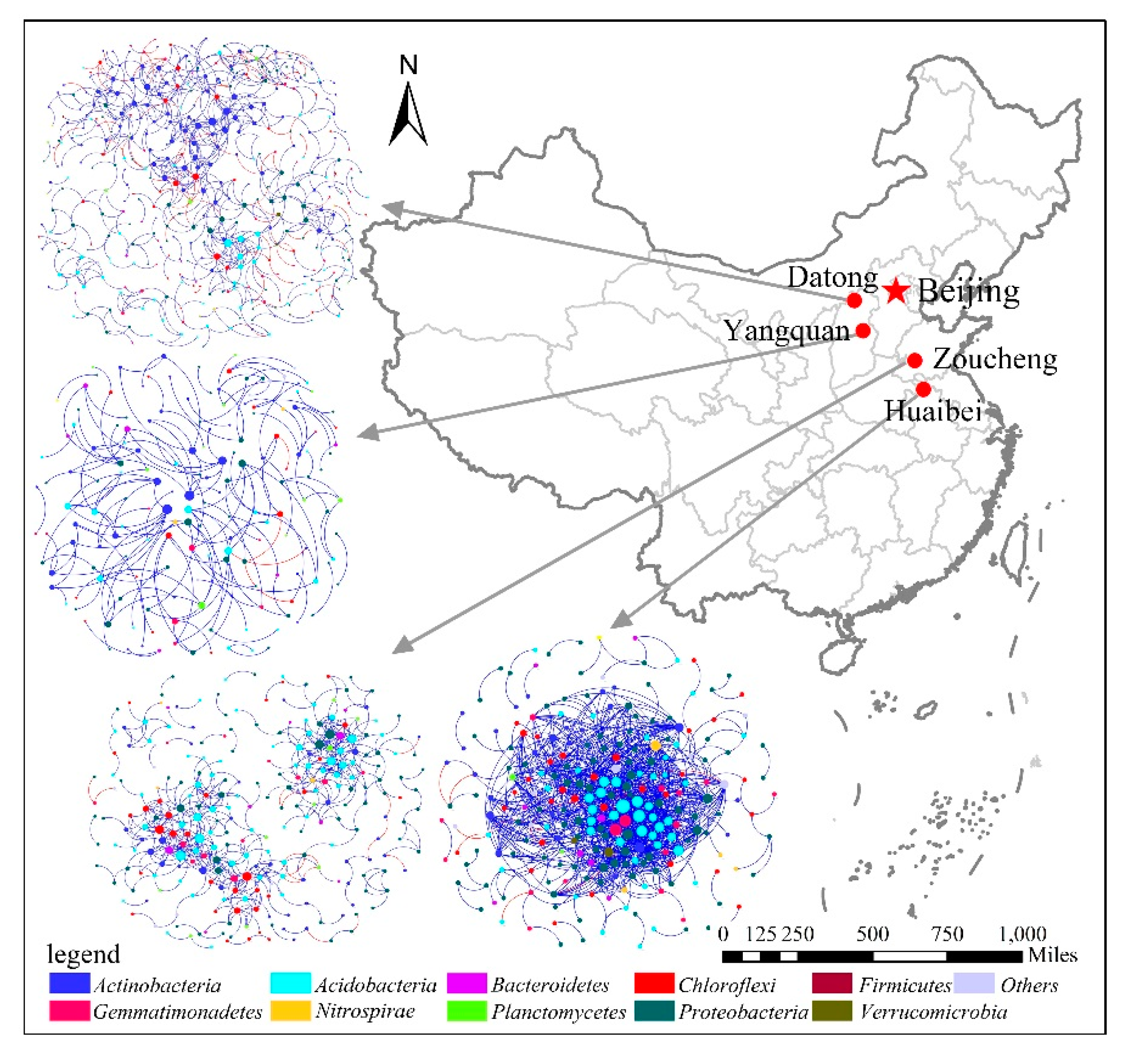
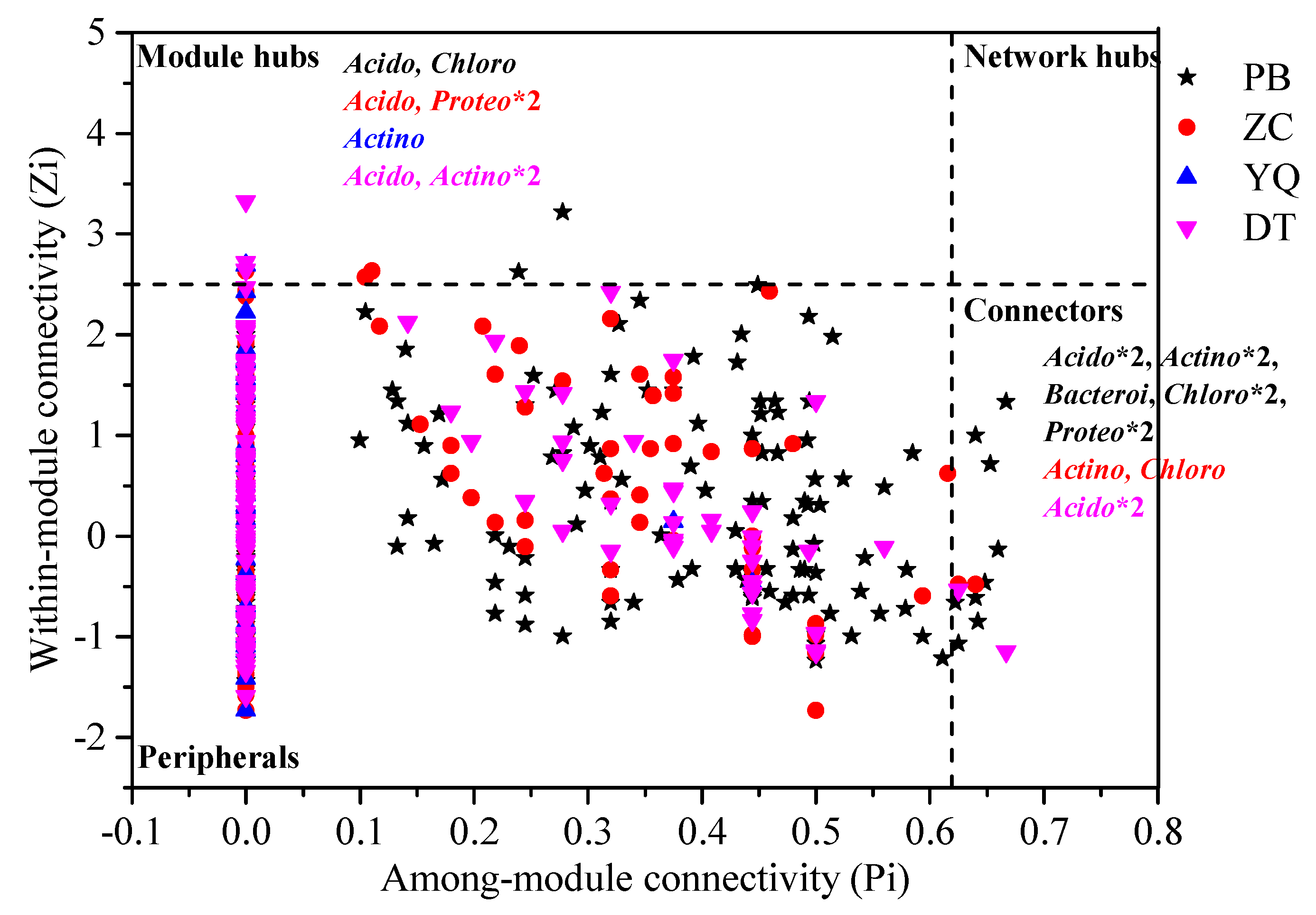
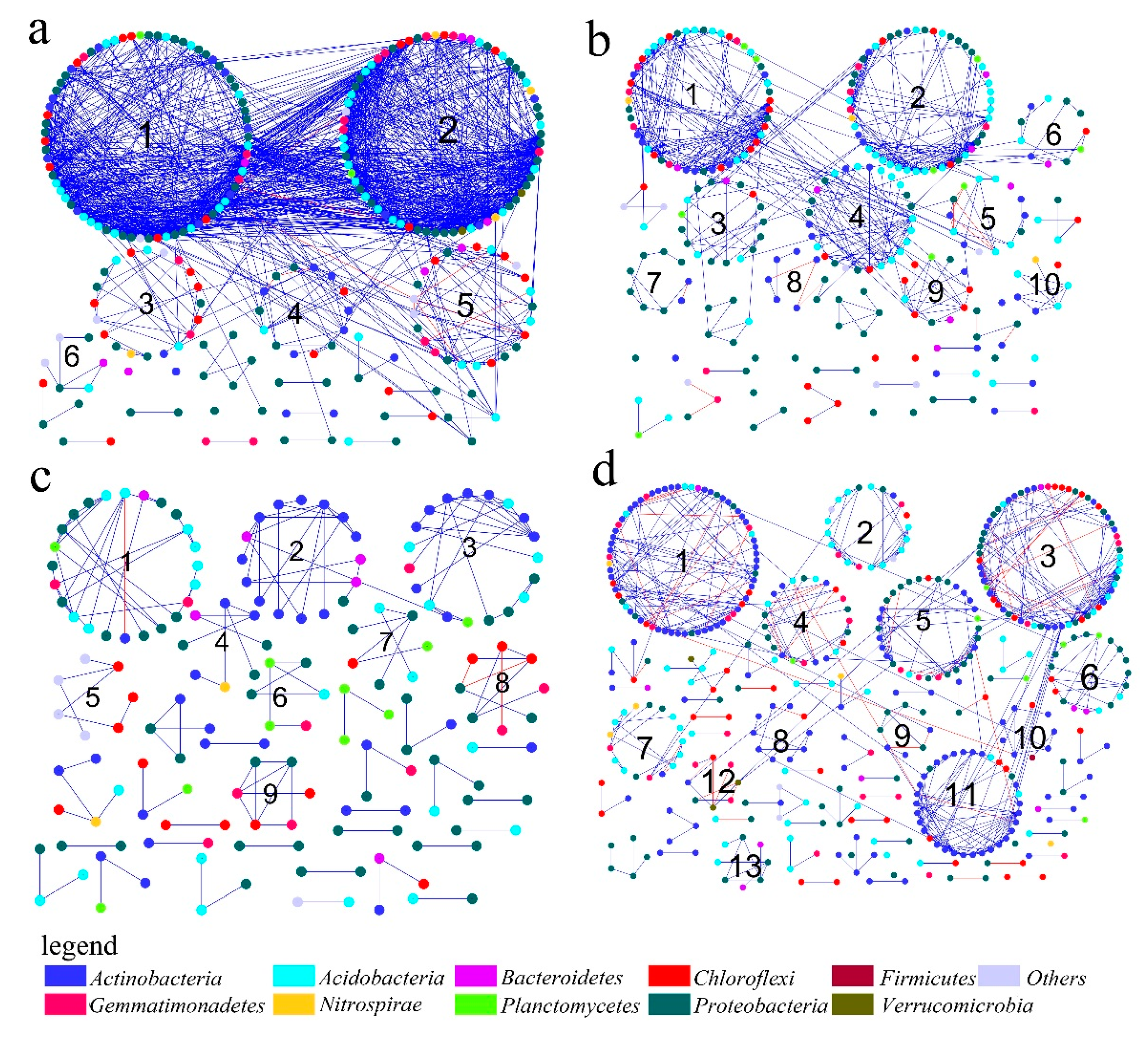
3.2. Topological Properties of MENs in Different Mining Areas
3.3. Dominant Microbial Taxa across Different Mining Areas
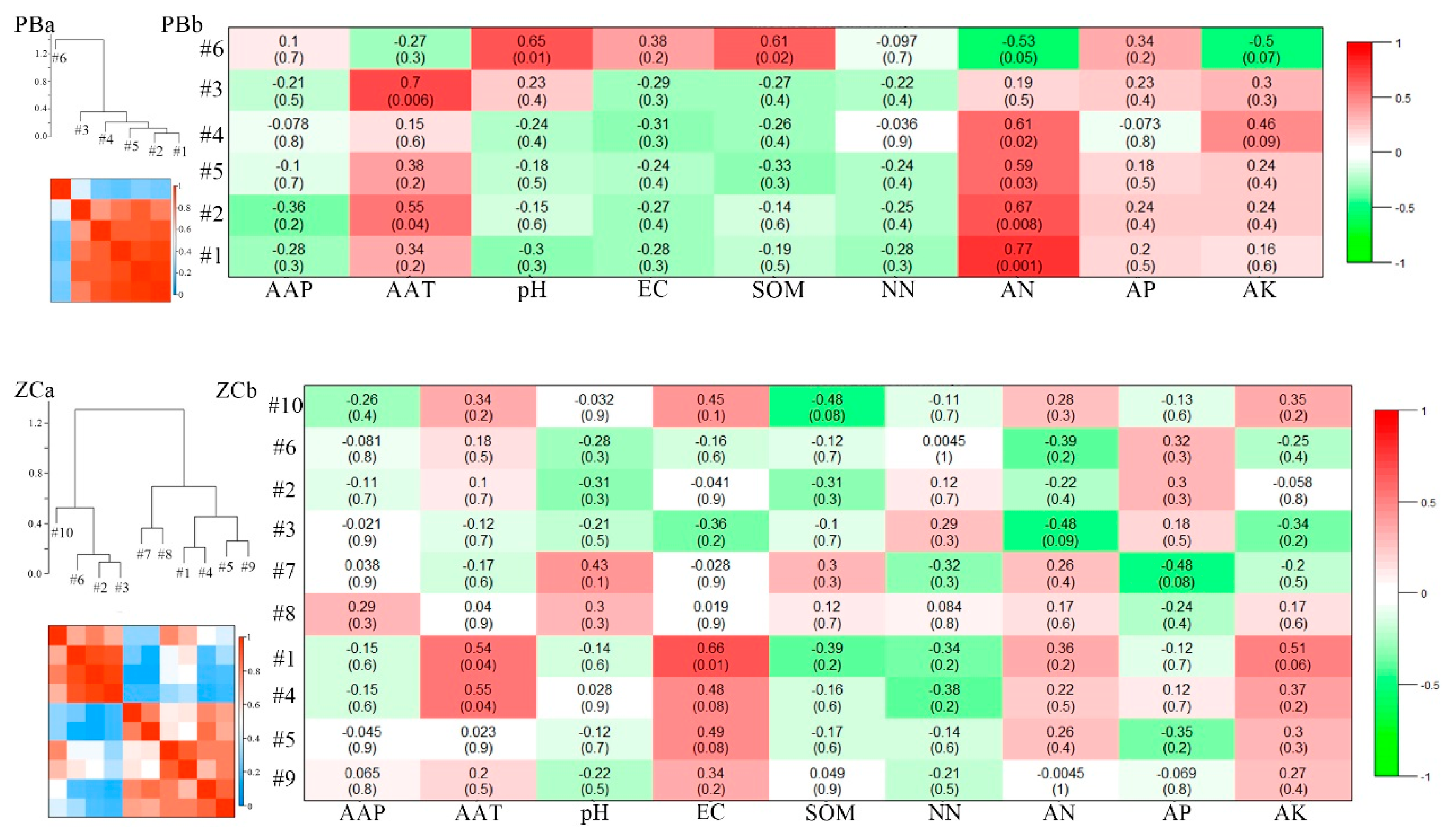
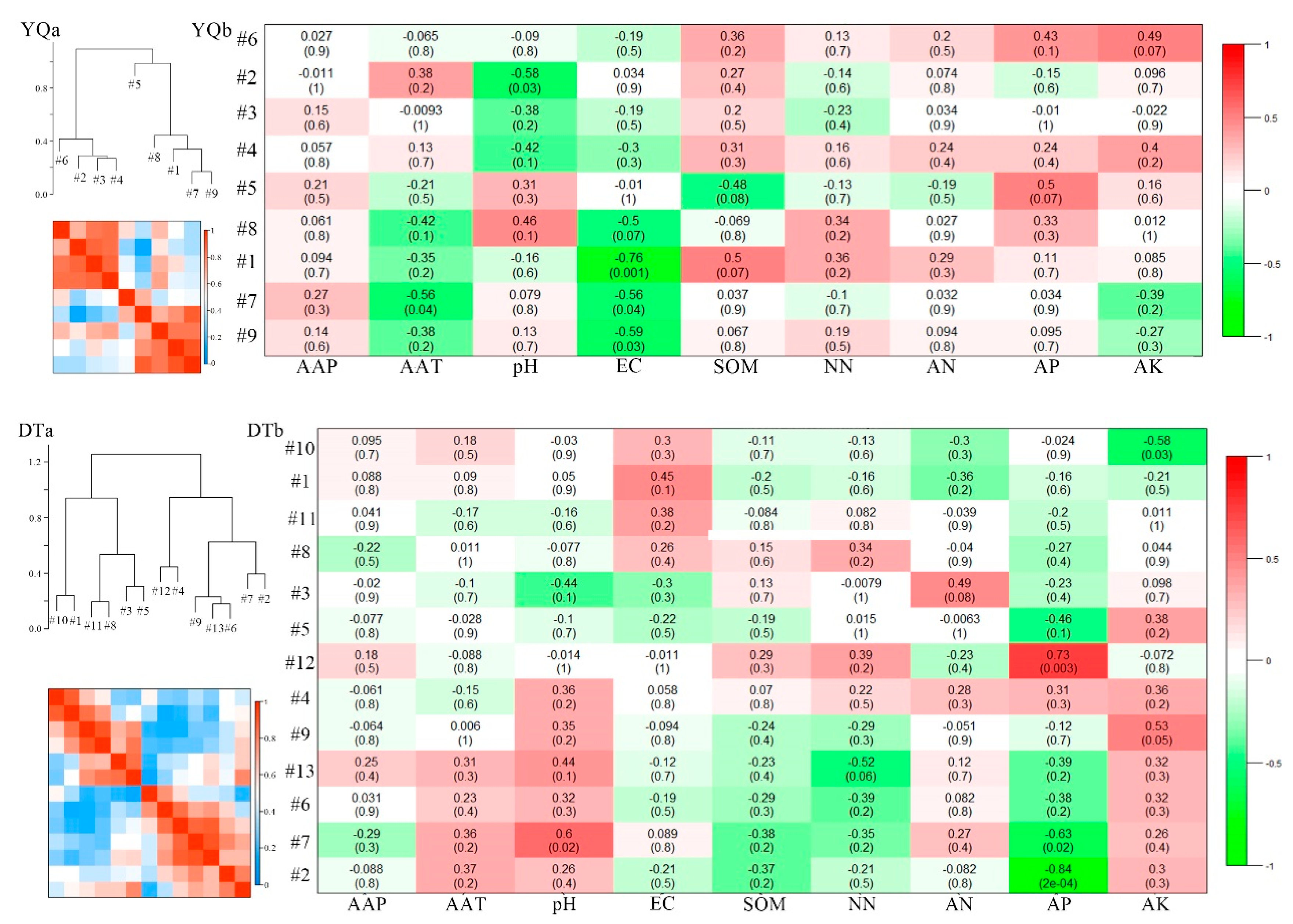
3.4. Eigengene Network Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| MEN | Molecular ecological network |
| PB | Peibei |
| ZC | Zoucheng |
| YQ | Yangquan |
| DT | Datong |
| PCA | Principal component analysis |
| NMDS | Non-metric multidimensional scaling |
| avgCC | Average clustering coefficient |
| avgK | Average degrees |
| GD | Average path distance |
| SRAs | Standardized relative abundances |
| AAP | Annual average precipitation |
| AAT | Annual average temperature |
| SOM | Soil organic matter |
| AN | Ammonium nitrate |
| EC | Electrical conductivity |
| NN | Nitrate-nitrogen |
| AP | Available phosphorus |
| AK | Available potassium |
| CCA | Canonical correspondence analysis |
| VPA | Variation partition analysis |
| RRC | Response ratio calculation |
| LEfSe | Linear discriminant analysis Effect Size |
References
- Bian, Z.; Inyang, H.I.; Daneils, J.L.; Otto, F.; Struthers, S. Environmental issues from coal mining and their solutions. Int. J. Min. Sci. Technol. 2010, 20, 215–223. [Google Scholar] [CrossRef]
- Hu, Z.; Chen, C.; Xiao, W.; Wang, X.; Gao, M. Surface movement and deformation characteristics due to high-intensive coal mining in the windy and sandy region. Int. J. Coal Sci. Technol. 2016, 3, 339–348. [Google Scholar] [CrossRef] [Green Version]
- Dangi, S.R.; Stahl, P.D.; Wick, A.F.; Ingram, L.J.; Buyer, J.S. Soil microbial community recovery in reclaimed soils on a surface coal mine site. Soil Sci. Soc. Am. J. 2012, 76, 915–924. [Google Scholar] [CrossRef]
- Li, J.; Zheng, Y.; Yan, J.; Li, H.; He, J. Succession of plant and soil microbial communities with restoration of abandoned land in the Loess Plateau, China. J. Soil Sediment. 2013, 13, 760–769. [Google Scholar] [CrossRef]
- Deng, H.; Li, X.F.; Cheng, W.D.; Zhu, Y.G. Resistance and resilience of Cu-polluted soil after Cu perturbation, tested by a wide range of soil microbial parameters. FEMS Microbiol. Ecol. 2009, 70, 137–148. [Google Scholar] [CrossRef] [Green Version]
- Novianti, V.; Marrs, R.H.; Choesin, D.N.; Iskandar, D.T.; Suprayogo, D. Natural regeneration on land degraded by coal mining in a tropical climate: Lessons for ecological restoration from Indonesia. Land Degrad. Dev. 2018, 29, 4050–4060. [Google Scholar] [CrossRef]
- Yuan, Y.; Zhao, Z.; Li, X.; Wang, Y.; Bai, Z. Characteristics of labile organic carbon fractions in reclaimed mine soils: Evidence from three reclaimed forests in the Pingshuo opencast coal mine, China. Sci. Total Environ. 2018, 613, 1196–1206. [Google Scholar] [CrossRef]
- Li, Y.; Chen, L.; Wen, H.; Zhou, T.; Zhang, T.; Gao, X. 454 Pyrosequencing analysis of bacterial diversity revealed by a comparative study of soils from mining subsidence and reclamation areas. J. Microbiol. Biotechnol. 2014, 24, 313–323. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Lei, S.; Gong, C. Comparison of plant and microbial communities between an artificial restoration and a natural restoration topsoil in coal mining subsidence area. Environ. Earth Sci. 2019, 78, 204. [Google Scholar] [CrossRef]
- Griffiths, B.S.; Philippot, L. Insights into the resistance and resilience of the soil microbial community. FEMS Microbiol. Rev. 2013, 37, 112–129. [Google Scholar] [CrossRef] [Green Version]
- Ladau, J.; Sharpton, T.J.; Finucane, M.M.; Jospin, G.; Kembel, S.W.; O’Dwyer, J.; Koeppel, A.F.; Green, J.L.; Pollard, K.S. Global marine bacterial diversity peaks at high latitudes in winter. ISME J. 2013, 7, 1669–1677. [Google Scholar] [CrossRef]
- Zhou, J.; Deng, Y.; Luo, F.; He, Z.; Tu, Q.; Zhi, X. Functional molecular ecological networks. MBio 2010, 4, e110–e169. [Google Scholar] [CrossRef] [Green Version]
- Morriën, E.; Hannula, S.E.; Snoek, L.B.; Helmsing, N.R.; Zweers, H.; de Hollander, M.; Soto, R.L.; Bouffaud, M.; Buée, M.; Dimmers, W.; et al. Soil networks become more connected and take up more carbon as nature restoration progresses. Nat. Commun. 2017, 8. [Google Scholar] [CrossRef]
- Mummey, D.L.; Stahl, P.D.; Buyer, J.S. Soil microbiological properties 20 years after surface mine reclamation: Spatial analysis of reclaimed and undisturbed sites. Soil Biol. Biochem. 2002, 34, 1717–1725. [Google Scholar] [CrossRef]
- Larsen, P.E.; Field, D.; Gilbert, J.A. Predicting bacterial community assemblages using an artificial neural network approach. Nat. Methods 2012, 9, 621–625. [Google Scholar] [CrossRef] [Green Version]
- Gazol, A.; Camarero, J.J.; Anderegg, W.R.L.; Vicente-Serrano, S.M. Impacts of droughts on the growth resilience of Northern Hemisphere forests. Glob. Ecol. Biogeogr. 2017, 26, 166–176. [Google Scholar] [CrossRef]
- Helingerová, M.; Frouz, J.; Šantrůčková, H. Microbial activity in reclaimed and unreclaimed post-mining sites near Sokolov (Czech Republic). Ecol. Eng. 2010, 36, 768–776. [Google Scholar] [CrossRef]
- Dimitriu, P.A.; Prescott, C.E.; Quideau, S.A.; Grayston, S.J. Impact of reclamation of surface-mined boreal forest soils on microbial community composition and function. Soil Biol. Biochem. 2010, 42, 2289–2297. [Google Scholar] [CrossRef]
- Pascual, J.A.; Garcia, C.; Hernandez, T.; Moreno, J.L.; Ros, M. Soil microbial activity as a biomarker of degradation and remediation processes. Soil Biol. Biochem. 2000, 32, 1877–1883. [Google Scholar] [CrossRef]
- Wang, S.; Brose, U. Biodiversity and ecosystem functioning in food webs the vertical diversity hypothesis. Ecol. Lett. 2018, 1, 9–20. [Google Scholar] [CrossRef]
- Deng, Y.; Zhang, P.; Qin, Y.; Tu, Q.; Yang, Y.; He, Z.; Schadt, C.W.; Zhou, J. Network succession reveals the importance of competition in response to emulsified vegetable oil amendment for uranium bioremediation. Environ. Microbiol. 2016, 1, 205–218. [Google Scholar] [CrossRef]
- Van Horn, D.J.; Wolf, C.R.; Colman, D.R.; Jiang, X.; Kohler, T.J.; McKnight, D.M.; Stanish, L.F.; Yazzie, T.; Takacs-Vesbach, C.D. Patterns of bacterial biodiversity in the glacial meltwater streams of the McMurdo Dry Valleys, Antarctica. FEMS Microbiol. Ecol. 2016, 92, w148. [Google Scholar] [CrossRef] [Green Version]
- Jiang, X.; Takacs-Vesbach, C.D. Microbial community analysis of pH 4 thermal springs in Yellowstone National Park. Extremophiles 2017, 21, 135–152. [Google Scholar] [CrossRef]
- Zhou, J.; Deng, Y.; Luo, F.; He, Z.; Yang, Y. Phylogenetic molecular ecological network of soil microbial communities in response to elevated CO2. MBio 2011, 2, e00122–e11. [Google Scholar] [CrossRef] [Green Version]
- Deng, Y.; Jiang, Y.H.; Yang, Y.; He, Z.; Luo, F.; Zhou, J. Molecular ecological network analyses. BMC Bioinform. 2012, 13, 113. [Google Scholar] [CrossRef] [Green Version]
- Doncheva, N.T.; Morris, J.H.; Gorodkin, J.; Jensen, L.J. Cytoscape StringApp: Network Analysis and Visualization of Proteomics Data. J. Proteome Res. 2018, 18, 623–632. [Google Scholar] [CrossRef]
- Wang, S.; Wang, X.; Han, X.; Deng, Y. Higher precipitation strengthens the microbial interactions in semi-arid grassland soils. Glob. Ecol. Biogeogr. 2018, 5, 570–580. [Google Scholar] [CrossRef]
- Layeghifard, M.; Hwang, D.M.; Guttman, D.S. Disentangling interactions in the microbiome: A network perspective. Trends Microbiol. 2017, 25, 217–228. [Google Scholar] [CrossRef]
- Li, X.X.; Zhang, Q.; Ma, J.; Yang, Y.J.; Wang, Y.F.; Fu, C. Flooding Irrigation Weakens the MolecularEcological Network Complexity of Soil Microbes During the Process of Dryland-to-Paddy Conversion. Int. J. Environ. Res. Public Health 2020, 17, 561. [Google Scholar] [CrossRef] [Green Version]
- Shendure, J.; Balasubramanian, S.; Church, G.M.; Gilbert, W.; Rogers, J.; Schloss, J.A.; Waterston, R.H. DNA sequencing at 40: Past, present and future. Nature 2017, 550, 345–353. [Google Scholar] [CrossRef]
- Wang, X.; Chen, G. Complex network:small-world, scale-free and beyond. IEEE Circ. Syst. Mag. 2003, 1, 6–20. [Google Scholar] [CrossRef] [Green Version]
- Hunt, D.E.; Ward, C.S. A network-based approach to disturbance transmission through microbial interactions. Front. Microbiol. 2015, 6, 1182. [Google Scholar] [CrossRef]
- Sheng, M.; Chen, X.; Zhang, X.; Hamel, C.; Cui, X.; Chen, J.; Chen, H.; Tang, M. Changes in arbuscular mycorrhizal fungal attributes along a chronosequence of black locust (Robinia pseudoacacia) plantations can be attributed to the plantation-induced variation in soil properties. Sci. Total Environ. 2017, 599–600. [Google Scholar] [CrossRef]
- Kitano, H. Biological robustness. Nat. Rev. Genet. 2004, 5, 826–837. [Google Scholar] [CrossRef]
- Delgado-Baquerizo, M.; Oliverio, A.M.; Brewer, T.E.; Benavent-González, A.; Eldridge, D.J.; Bardgett, R.D.; Maestre, F.T.; Singh, B.K.; Fierer, N. A global atlas of the dominant bacteria found in soil. Science 2018, 359, 320–325. [Google Scholar] [CrossRef] [Green Version]
- Berry, D.; Widder, S. Deciphering microbial interactions and detecting keystone species with co-occurrence networks. Front. Microbiol. 2014, 5. [Google Scholar] [CrossRef] [Green Version]
- Fierer, N. Embracing the unknown: Disentangling the complexities of the soil microbiome. Nat. Rev. Microbiol. 2017, 15, 579–590. [Google Scholar] [CrossRef]
- Ramirez, K.S.; Geisen, S.; Morriën, E.; Snoek, B.L.; van der Putten, W.H. Network analyses can advance above-belowground ecology. Trends Plant Sci. 2018, 23, 759–768. [Google Scholar] [CrossRef]
- Banerjee, S.; Walder, F.; Büchi, L.; Meyer, M.; Held, A.Y.; Gattinger, A.; Keller, T.; Charles, R.; van der Heijden, M.G.A. Agricultural intensification reduces microbial network complexity and the abundance of keystone taxa in roots. ISME J. 2019, 13, 1722–1736. [Google Scholar] [CrossRef] [Green Version]
- Ma, J.; Zhang, W.; Zhang, S.; Zhu, Q.; Feng, Q.; Chen, F. Short-term effects of CO2 leakage on the soil bacterial community in a simulated gas leakage scenario. PeerJ 2017, 5, e4024. [Google Scholar] [CrossRef] [Green Version]
- Tringe, S.G.; von Mering, C.; Kobayashi, A.; Salamov, A.A.; Chen, K.; Chang, H.W.; Podar, M.; Short, J.M.; Mathur, E.J.; Detter, J.C.; et al. Comparative metagenomics of microbial communities. Science 2005, 5721, 554–557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Sui, Y.; Yu, Z.; Yao, Q.; Shi, Y.; Chu, H.; Jin, J.; Liu, X.; Wang, G. Diversity and distribution patterns of acidobacterial communities in the black soil zone of northeast China. Soil Biol. Biochem. 2016, 95, 212–222. [Google Scholar] [CrossRef]
- Chen, F.; Zhang, W.; Ma, J.; Yang, Y.; Zhang, S.; Chen, R. Experimental study on the effects of underground CO2 leakage on soil microbial consortia. Int. J. Greenh. Gas. Con. 2017, 63, 241–248. [Google Scholar] [CrossRef]
- Zeng, Y.; Baumbach, J.; Barbosa, E.G.V.; Azevedo, V.; Zhang, C.; Koblížek, M. Metagenomic evidence for the presence of phototrophic Gemmatimonadetes bacteria in diverse environments. Environ. Microbiol. Rep. 2016, 1, 139–149. [Google Scholar] [CrossRef]
- Park, D.; Kim, H.; Yoon, S. Nitrous oxide reduction by an obligate aerobic bacterium, Gemmatimonas aurantiaca strain T-27. Appl. Environ. Microbiol. 2017, 12, e502–e517. [Google Scholar] [CrossRef] [Green Version]
- Tobin-Janzen, T.; Shade, A.; Marshall, L.; Torres, K.; Beblo, C.; Janzen, C.; Lenig, J.; Martinez, A.; Ressler, D. Nitrogen changes and domain bacteria ribotype diversity in soils overlying the Centralia, Pennsylvania underground coal mine fire. Soil Sci. 2005, 3, 191–201. [Google Scholar] [CrossRef]
- Sun, S.Y.; Sun, H.; Zhang, D.; Zhang, J.; Cai, Z.; Qin, G.; Song, Y. Response of soil microbes to vegetation restoration in coal mining subsidence areas at Huaibei coal mine, China. Int. J. Environ. Res. Public Health 2019, 16, 1757. [Google Scholar] [CrossRef] [Green Version]
- Ezeokoli, O.T.; Mashigo, S.K.; Paterson, D.G.; Bezuidenhout, C.C.; Adeleke, R.A. Microbial community structure and relationship with physicochemical properties of soil stockpiles in selected South African opencast coal mines. Soil Sci. Plant. Nutr. 2019, 65, 332–341. [Google Scholar] [CrossRef]
- Chaffron, S.; Rehrauer, H.; Pernthaler, J.; von Mering, C. A global network of coexisting microbes from environmental and whole-genome sequence data. Genome Res. 2010, 20, 947–959. [Google Scholar] [CrossRef] [Green Version]
- Harris, J. Soil microbial communities and restoration ecology: Facilitators or followers? Science 2009, 325, 573–574. [Google Scholar] [CrossRef]
- Chen, F.; Yang, Y.; Ma, Y.; Hou, H.; Zhang, S.; Ma, J. Effects of CO2 leakage on soil bacterial communities from simulated CO2-EOR areas. Environ. Sci. Proc. Imp. 2016, 18, 547–554. [Google Scholar] [CrossRef]
- O’Brien, F.J.M.; Almaraz, M.; Foster, M.A.; Hill, A.F.; Huber, D.P.; King, E.K.; Langford, H.; Lowe, M.; Mickan, B.S.; Miller, V.S.; et al. Soil salinity and pH drive soil bacterial community composition and diversity along a lateritic slope in the Avon River critical zone observatory, Western Australia. Front. Microbiol. 2019, 10, 1486. [Google Scholar] [CrossRef]
- Zeng, Q.; An, S.; Liu, Y.; Wang, H.; Wang, Y. Biogeography and the driving factors affecting forest soil bacteria in an arid area. Sci. Total Environ. 2019, 680, 124–131. [Google Scholar] [CrossRef]
- Xiao, L.; Bi, Y.; Du, S.; Wang, Y.; Guo, C. Effects of re-vegetation type and arbuscular mycorrhizal fungal inoculation on soil enzyme activities and microbial biomass in coal mining subsidence areas of Northern China. Catena 2019, 177, 202–209. [Google Scholar] [CrossRef]
- Pille da Silva, E.; Dutra de Armas, R.; Ademar Avelar Ferreira, P.; Laurentino Dantas, M.K.; Giachini, A.J.; Rocha-Nicoleite, E.; Gonzalez, A.H.; Fonseca Sousa Soares, C.R. Soil attributes in coal mining areas under recovery with bracatinga (Mimosa scabrella). Lett. Appl. Microbiol. 2019, 68, 97–504. [Google Scholar] [CrossRef]
- Bi, Y.; Xiao, L.; Sun, J. An arbuscular mycorrhizal fungus ameliorates plant growth and hormones after moderate root damage due to simulated coal mining subsidence: A microcosm study. Environ. Sci. Pollut. Res. 2019, 26, 11053–11061. [Google Scholar] [CrossRef]
- Bi, Y.; Xie, L.; Wang, J.; Zhang, Y.; Wang, K. Impact of host plants, slope position and subsidence on arbuscular mycorrhizal fungal communities in the coal mining area of north-central China. J. Arid Environ. 2019, 163, 68–76. [Google Scholar] [CrossRef]
- Fernández-Montiel, I.; Touceda, M.; Pedescoll, A.; Gabilondo, R.; Prieto-Fernández, A.; Bécares, E. Short-term effects of simulated below-ground carbon dioxide leakage on a soil microbial community. Int. J. Greenhouse Gas Control 2015, 36, 51–59. [Google Scholar] [CrossRef]
- Li, J.; Xin, Z.; Yan, J.; Li, H.; Chen, J.; Ding, G. Physicochemical and microbiological assessment of soil quality on a chronosequence of a mine reclamation site. Eur. J. Soil Sci. 2018, 6, 1056–1067. [Google Scholar] [CrossRef]
- Sáenz De Miera, L.E.; Arroyo, P.; de Luis Calabuig, E.; Falagán, J.; Ansola, G. High-throughput sequencing of 16S RNA genes of soil bacterial communities from a naturally occurring CO2 gas vent. Int. J. Greenhouse Gas Control 2014, 29, 176–184. [Google Scholar] [CrossRef]
- Ngugi, M.R.; Dennis, P.G.; Neldner, V.J.; Doley, D.; Fechner, N.; McElnea, A. Open-cut mining impacts on soil abiotic and bacterial community properties as shown by restoration chronosequence. Restor. Ecol. 2018, 5, 839–850. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
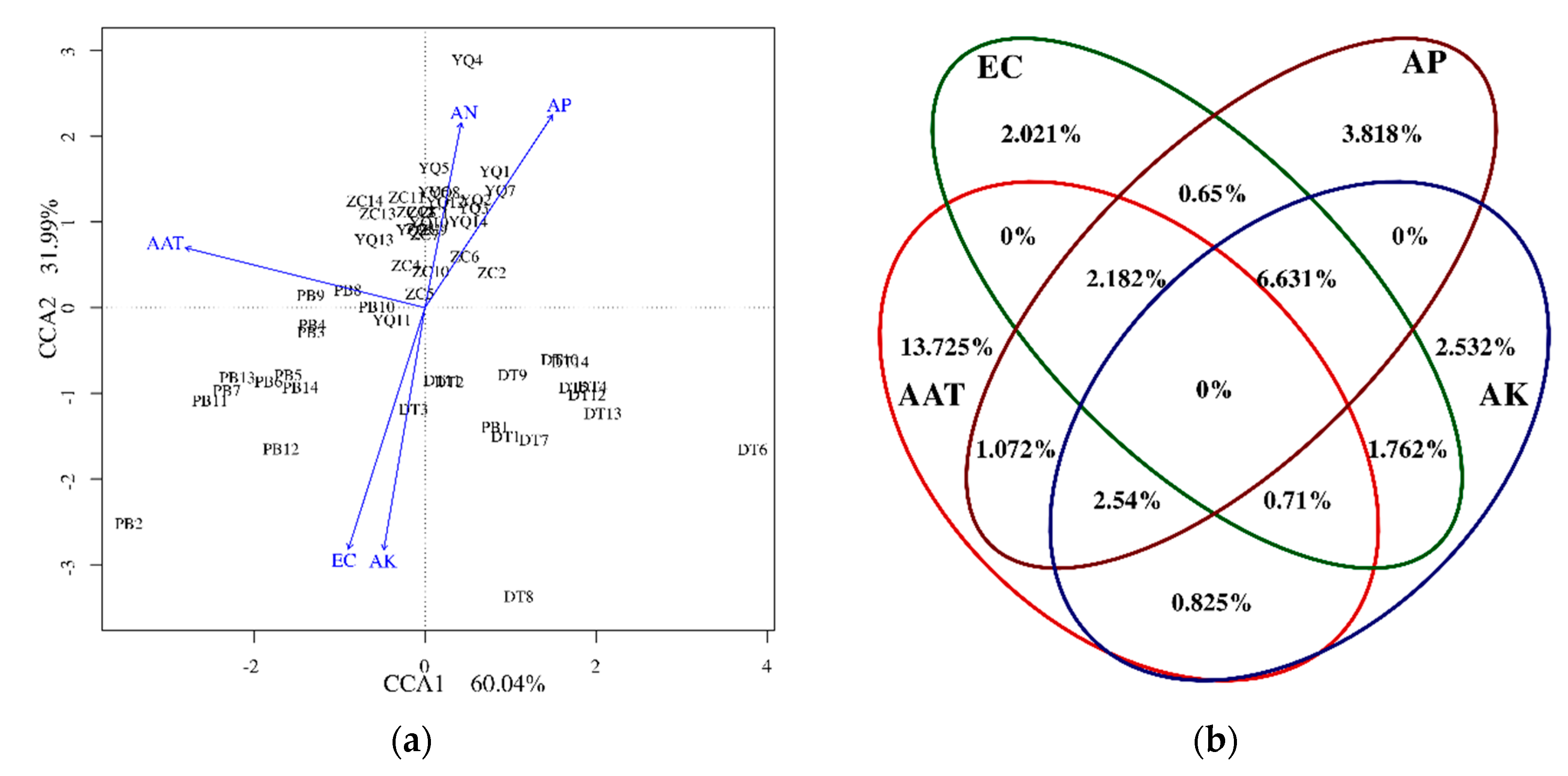{kind=link}
| Mining Area | Chao | Shannon | Pielou Evenness |
|---|---|---|---|
| PB | 3710.10 ± 836.20 b | 6.5937 ± 0.5738 ab | 0.8305 ± 0.0441 c |
| ZC | 8319.38 ± 541.91 ab | 7.7519 ± 0.0824 c | 0.9014 ± 0.0105 bc |
| YQ | 4917.14 ± 891.56 c | 6.8408 ± 0.2617 bc | 0.8463 ± 0.0177 ab |
| DT | 5238.99 ± 421.43 bc | 6.7701 ± 0.1585 b | 0.8120 ± 0.0144 ab |
| Network Indexes | PB | ZC | YQ | DT | |
|---|---|---|---|---|---|
| Empirical networks | Similarity threshold | 0.86 | 0.86 | 0.86 | 0.86 |
| R2 of power law | 0.837 | 0.931 | 0.852 | 0.896 | |
| Total nodes | 248 | 265 | 165 | 441 | |
| Total links | 1285 | 516 | 163 | 640 | |
| Average degree (avgK) | 10.363 | 3.894 | 1.976 | 2.902 | |
| Average clustering coefficient (avgCC) | 0.314 | 0.258 | 0.158 | 0.184 | |
| Average path distance (GD) | 3.334 | 7.725 | 3.975 | 7.802 | |
| Modularity | 0.364 | 0.701 | 0.897 | 0.829 | |
| Module number (with >5 nodes) | 6 | 10 | 9 | 13 | |
| Random networks | Average clustering coefficient (avgCC) | 0.134 ± 0.010 | 0.028 ± 0.006 | 0.007 ± 0.005 | 0.008 ± 0.003 |
| Average path distance (GD) | 2.772 ± 0.024 | 3.877 ± 0.058 | 6.454 ± 0.448 | 5.022 ± 0.076 | |
| Modularity | 0.228 ± 0.005 | 0.496 ± 0.008 | 0.795 ± 0.011 | 0.637 ± 0.008 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ma, J.; Lu, Y.; Chen, F.; Li, X.; Xiao, D.; Wang, H. Molecular Ecological Network Complexity Drives Stand Resilience of Soil Bacteria to Mining Disturbances among Typical Damaged Ecosystems in China. Microorganisms 2020, 8, 433. https://doi.org/10.3390/microorganisms8030433
Ma J, Lu Y, Chen F, Li X, Xiao D, Wang H. Molecular Ecological Network Complexity Drives Stand Resilience of Soil Bacteria to Mining Disturbances among Typical Damaged Ecosystems in China. Microorganisms. 2020; 8(3):433. https://doi.org/10.3390/microorganisms8030433
Chicago/Turabian StyleMa, Jing, Yongqiang Lu, Fu Chen, Xiaoxiao Li, Dong Xiao, and Hui Wang. 2020. "Molecular Ecological Network Complexity Drives Stand Resilience of Soil Bacteria to Mining Disturbances among Typical Damaged Ecosystems in China" Microorganisms 8, no. 3: 433. https://doi.org/10.3390/microorganisms8030433