Epstein–Barr Virus Infection of Oral Squamous Cells

 , ,
, ,  and
and
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Patient Specimens
2.2. Cell Culture
2.3. EBV Detection by PCR and EBER in Situ Hybridization (ISH)
2.4. Establishment of EBV-Positive SCC Cells
2.5. Western Blotting
2.6. Quantitative Reverse Transcription PCR (qRT-PCR)
2.7. Quantification of EBV Copy Number
2.8. Immunofluorescence Staining
2.9. Virus Titration by Flow Cytometry
2.10. Cell Proliferation Assay
2.11. Wound Healing Assay
2.12. Cell Invasion and Migration Assay
2.13. Apoptosis Assay
2.14. Statistical Analysis
3. Results
3.1. Detection of EBV-Positive Cells in OSCC Tissues
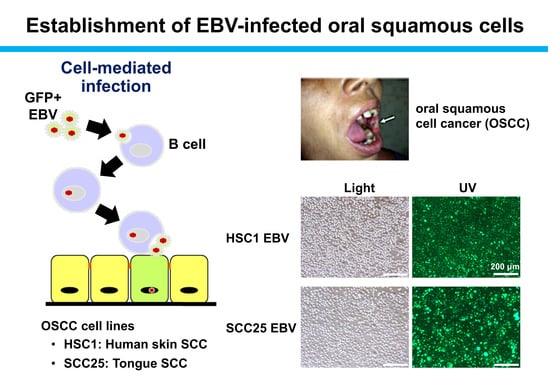
3.2. Establishment of EBV-Infected SCC Cells
3.3. Induction of Lytic EBV Replication in Latently Infected SCC Cells
3.4. EBV Infection Promotes Cell Proliferation and Inhibits Programmed-Cell Death in SCC Cells.
3.5. EBV Induces Mesenchymal Properties of OSCC Cells
3.6. EBV Promotes Migration and Invasion in SCC Cells.
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Longnecker, R.M.; Kieff, E.; Cohen, J.I. Epstein-Barr virus. In Fields Virology, 6th ed.; Fields, B.N., Knipe, D.M., Howley, P.M., Eds.; Lippincott-Williams & Wilkins: Philadelphia, PA, USA, 2013; pp. 1898–1959. [Google Scholar]
- Vockerodt, M.; Yap, L.F.; Shannon-Lowe, C.; Curley, H.; Wei, W.; Vrzalikova, K.; Murray, P.G. The Epstein-Barr virus and the pathogenesis of lymphoma. J. Pathol. 2015, 235, 312–322. [Google Scholar] [CrossRef] [PubMed]
- Tsao, S.W.; Tsang, C.M.; To, K.F.; Lo, K.W. The role of Epstein-Barr virus in epithelial malignancies. J. Pathol. 2015, 235, 323–333. [Google Scholar] [CrossRef] [PubMed]
- Iizasa, H.; Nanbo, A.; Nishikawa, J.; Jinushi, M.; Yoshiyama, H. Epstein-Barr Virus (EBV)-associated gastric carcinoma. Viruses 2012, 4, 3420–3439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allen, C.; Rooney, C.M.; Gottschalk, S. Infectious Mononucleosis and other Epstein-Barr Virus–associated Diseases. In Hematology, 7th ed.; Elsevier: Philadelphia, PA, USA, 2018; pp. 747–759. [Google Scholar]
- Neville, B.W.; Damm, D.D.; Allen, C.M. Oral and Maxillofacial Pathology, 4th ed.; Elsevier Health Sciences: St. Louis, MO, USA, 2015; pp. 241–242. [Google Scholar]
- Acharya, S.; Ekalaksananan, T.; Vatanasapt, P.; Loyha, K.; Phusingha, P.; Promthet, S.; Kongyingyoes, B.; Pientong, C. Association of Epstein-Barr virus infection with oral squamous cell carcinoma in a case-control study. J. Oral. Pathol. Med. 2015, 44, 252–257. [Google Scholar] [CrossRef] [PubMed]
- Higa, M.; Kinjo, T.; Kamiyama, K.; Chinen, K.; Iwamasa, T.; Arasaki, A.; Sunakawa, H. Epstein-Barr virus (EBV)-related oral squamous cell carcinoma in Okinawa, a subtropical island, in southern Japan--simultaneously infected with human papillomavirus (HPV). Oral. Oncol. 2003, 39, 405–414. [Google Scholar] [CrossRef]
- Deng, Z.; Uehara, T.; Maeda, H.; Hasegawa, M.; Matayoshi, S.; Kiyuna, A.; Agena, S.; Pan, X.; Zhang, C.; Yamashita, Y.; et al. Epstein-Barr virus and human papillomavirus infections and genotype distribution in head and neck cancers. Plos ONE 2014, 9, e113702. [Google Scholar] [CrossRef]
- Drop, B.; Strycharz-Dudziak, M.; Kliszczewska, E.; Polz-Dacewicz, M. Coinfection with Epstein-Barr virus (EBV), human papilloma virus (HPV) and polyoma BK virus (BKPyV) in laryngeal, oropharyngeal and oral cavity cancer. Int. J. Mol. Sci. 2017, 18, E2752. [Google Scholar] [CrossRef] [Green Version]
- Yen, C.-Y.; Lu, M.-C.; Tzeng, C.-C.; Huang, J.-Y.; Chang, H.-W.; Chen, R.-S.; Liu, S.-Y.; Liu, S.-T.; Shieh, B.; Li, C. Detection of EBV infection and gene expression in oral cancer from patients in Taiwan by microarray analysis. J. Biomed. Biotechnol. 2009, 2009, 904589. [Google Scholar] [CrossRef]
- Kikuchi, K.; Noguchi, Y.; de Rivera, M.W.G.-N.; Hoshino, M.; Sakashita, H.; Yamada, T.; Inoue, H.; Miyazaki, Y.; Nozaki, T.; Silvia, B. Detection of Epstein-Barr virus genome and latent infection gene expression in normal epithelia, epithelial dysplasia, and squamous cell carcinoma of the oral cavity. Tumour. Biol. 2016, 37, 3389–3404. [Google Scholar] [CrossRef]
- She, Y.; Nong, X.; Zhang, M.; Wang, M. Epstein-Barr virus infection and oral squamous cell carcinoma risk: a meta-analysis. PLoS ONE 2017, 12, e0186860. [Google Scholar]
- Young, L.S.; Yap, L.F.; Murray, P.G. Epstein–Barr virus: more than 50 years old and still providing surprises. Nat. Rev. Cancer. 2016, 16, 789–802. [Google Scholar] [CrossRef] [PubMed]
- Pathmanathan, R.; Prasad, U.; Sadler, R.; Flynn, K.; Raab-Traub, N. Clonal proliferations of cells infected with Epstein–Barr virus in preinvasive lesions related to nasopharyngeal carcinoma. N Engl. J. Med. 1995, 333, 693–698. [Google Scholar] [CrossRef] [PubMed]
- Tsao, S.W.; Tsang, C.M.; Lo, K.W. Epstein-Barr virus infection and nasopharyngeal carcinoma. Philos Trans. R Soc. Lond. B Biol. Sci. 2017, 372, 20160270. [Google Scholar] [CrossRef] [PubMed]
- Raab-Traub, N.; Flynn, K. The structure of the termini of the Epstein-Barr virus as a marker of clonal cellular proliferation. Cell. 1986, 47, 883–889. [Google Scholar] [CrossRef]
- Imai, S.; Koizumi, S.; Sugiura, M.; Tokunaga, M.; Uemura, Y.; Yamamoto, N.; Tanaka, S.; Sato, E.; Osato, T. Gastric carcinoma: monoclonal epithelial malignant cells expressing Epstein-Barr virus latent infection protein. Proc. Natl. Acad. Sci. USA. 1994, 91, 9131–9135. [Google Scholar] [CrossRef] [Green Version]
- Maruo, S.; Yang, L.; Takada, K. Roles of Epstein-Barr virus glycoproteins gp350 and gp25 in the infection of human epithelial cells. J. Gen. Virol. 2001, 82, 2373–2383. [Google Scholar] [CrossRef]
- Peferoen, L.A.; Lamers, F.; Lodder, L.N.; Gerritsen, W.H.; Huitinga, I.; Melief, J.; Giovannoni, G.; Meier, U.; Hintzen, R.Q.; Verjans, G.M.G.M.; et al. Epstein Barr virus is not a characteristic feature in the central nervous system in established multiple sclerosis. Brain 2010, 133, e137. [Google Scholar] [CrossRef] [Green Version]
- Imai, S.; Nishikawa, J.; Takada, K. Cell-to-cell contact as an efficient mode of Epstein-Barr virus infection of diverse human epithelial cells. J. Virol. 1998, 72, 4371–4378. [Google Scholar] [CrossRef] [Green Version]
- Le, Q.T.; Jones, C.D.; Yau, T.K.; Shirazi, H.A.; Wong, P.H.; Thomas, E.N.; Patterson, B.K.; Lee, A.W.N.; Zehnder, J.L. A comparison study of different PCR assays in measuring circulating plasma Epstein-Barr virus DNA levels in patients with nasopharyngeal carcinoma. Clin. Cancer Res. 2005, 11, 5700–5707. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Maruo, S.; Takada, K. CD21-mediated entry and stable infection by Epstein-Barr virus in canine and rat cells. J. Virol. 2000, 74, 10745–10751. [Google Scholar] [CrossRef] [Green Version]
- Katsumura, K.R.; Maruo, S.; Wu, Y.; Kanda, T.; Takada, K. Quantitative evaluation of the role of Epstein-Barr virus immediate-early protein BZLF1 in B-cell transformation. J. Gen. Virol. 2009, 90, 2331–2341. [Google Scholar] [CrossRef] [PubMed]
- Chaffer, C.L.; Weinberg, R.A. A perspective on cancer cell metastasis. Science 2011, 331, 1559–1564. [Google Scholar] [CrossRef] [PubMed]
- Birdwell, C.E.; Queen, K.J.; Kilgore, P.C.; Rollyson, P.; Trutschl, M.; Cvek, U.; Scott, R.S. Genome-wide DNA methylation as an epigenetic consequence of Epstein-Barr virus infection of immortalized keratinocytes. J. Virol. 2014, 88, 11442–11458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eichelberg, M.R.; Welch, R.; Guidry, J.T.; Ali, A.; Ohashi, M.; Makielski, K.R.; McChesney, K.; Sciver, N.V.; Lambert, P.F.; Keles, S.; et al. Epstein-Barr virus infection promotes epithelial cell growth by attenuating differentiation-dependent exit from the cell cycle. mBio 2019, 10, e01332. [Google Scholar] [CrossRef] [Green Version]
- Pegtel, D.M.; Middeldorp, J.; Thorley-Lawson, D.A. Epstein-Barr virus infection in ex vivo tonsil epithelial cell cultures of asymptomatic carriers. J. Virol. 2004, 78, 12613–12624. [Google Scholar] [CrossRef] [Green Version]
- Nawandar, D.M.; Wang, A.; Makielski, K.; Lee, D.; Ma, S.; Barlow, E.; Reusch, J.; Jiang, R.; Wille, C.K.; Greenspan, D.; et al. Differentiation-dependent KLF4 expression promotes lytic Epstein-Barr virus infection in epithelial cells. PLoS Pathog. 2015, 11, e1005195. [Google Scholar] [CrossRef]
- Murata, T.; Narita, Y.; Sugimoto, A.; Kawashima, D.; Kanda, T.; Tsurumi, T. Contribution of myocyte enhancer factor 2 (MEF2) family transcription factors to BZLF1 expression in Epstein-Barr virus reactivation from latency. J. Virol. 2013, 87, 10148–10162. [Google Scholar] [CrossRef] [Green Version]
- Murata, T. Regulation of Epstein–Barr virus reactivation from latency. Microbiol. Immunol. 2014, 58, 307–317. [Google Scholar] [CrossRef]
- Kraus, R.J.; Yu, X.; Blue-leaf, A.C.; Sathiamoorthi, S.; Iempridee, T.; Nawandar, D.M.; Ma, S.; Romero-Masters, J.C.; McChesney, K.G.; Lin, Z.; et al. Hypoxia-inducible factor-1α plays roles in Epstein-Barr virus’s natural life cycle and tumorigenesis by inducing lytic infection through direct binding to the immediate-early BZLF1 gene promoter. PLoS Pathog. 2017, 13, e1006404. [Google Scholar] [CrossRef]
- Lu, J.; Tang, M.; Li, H.; Xu, Z.; Weng, X.; Li, J.; Yu, X.; Zhao, L.; Liu, H.; Hu, Y.; et al. EBV-LMP1 suppresses the DNA damage response through DNA-PK/AMPK signaling to promote radioresistance in nasopharyngeal carcinoma. Cancer Lett. 2016, 380, 191–200. [Google Scholar] [CrossRef]
- Li, L.; Li, W.; Xiao, L.; Xu, J.; Chen, X.; Tang, M.; Dong, Z.; Tao, Q.; Cao, Y. Viral oncoprotein LMP1 disrupts p53-induced cell cycle arrest and apoptosis through modulating K63-linked ubiquitination of p53. Cell Cycle. 2012, 11, 2327–2336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, G.-D.; Huang, T.-J.; Peng, L.-X.; Yang, C.-F.; Liu, R.-Y.; Huang, H.-B.; Chu, Q.-Q.; Yang, H.-J.; Huang, J.-L.; Zhu, Z.-Y.; et al. Epstein-Barr virus encoded LMP1 upregulates microRNA-21 to promote the resistance of nasopharyngeal carcinoma cells to cisplatin-induced apoptosis by suppressing PDCD4 and Fas-L. PLoS ONE 2013, 8, e78355. [Google Scholar]
- Kang, D.; Skalsky, R.-L.; Cullen, B.-R. EBV BART microRNAs target multiple pro-apoptotic cellular genes to promote epithelial cell survival. PLoS Pathog. 2015, 11, e1004979. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Burassakarn, A.; Kang, Y.; Iizasa, Y.; Yoshiyama, H. A single nucleotide polymorphism in BART promoter region of Epstein-Barr virus isolated from nasopharyngeal cancer cells. Biochem. Biophys Res. Com. 2019, 520, 373–378. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Han, T.; Sheng, X.; Zhang, N.; Wang, P. Downregulation of EB virus miR-BART4 inhibits proliferation and aggressiveness while promoting radiosensitivity of nasopharyngeal carcinoma. Biomed. Pharmacother 2018, 108, 741–751. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Wang, J.; Wei, L.; Peng, Q.; Gao, Y.; Fu, Y.; Lu, Y.; Qin, Z.; Zhang, X.; Lu, J.; et al. Epstein-Barr virus microRNA miR-BART5-3p inhibits p53 expression. J. Virol. 2018, 92, e0102. [Google Scholar] [CrossRef] [Green Version]
- Cheng, T.-C.; Hsieh, S.-S.; Hsu, W.-L.; Chen, Y.-F.; Ho, H.-H.; Sheu, L.-F. Expression of Epstein-Barr nuclear antigen 1 in gastric carcinoma cells is associated with enhanced tumorigenicity and reduced cisplatin sensitivity. Int. J. Oncol. 2010, 36, 151–160. [Google Scholar]
- Wang, K.; Ge, Y.; Ni, C.; Cui, B.; Du, J.; Zhang, B. Epstein-Barr virus-induced up-regulation of TCAB1 is involved in the DNA damage response in nasopharyngeal carcinoma. Sci. Rep. 2017, 7, 3218. [Google Scholar] [CrossRef] [Green Version]
- Yang, F.; Liu, Q.; Hu, C. Epstein-Barr virus-encoded LMP1 increases miR-155 expression, which promotes radioresistance of nasopharyngeal carcinoma via suppressing UBQLN1. Eur. Rev. Med. Pharmacol. Sci. 2015, 19, 4507–4515. [Google Scholar]
- Zuo, L.; Zhang, J.; Liu, L.; Zhou, Q.; Du, S.; Xin, S.; Ning, Z.-P.; Yang, J.; Yu, H.-B.; Yue, W.-X.; et al. Cadherin 6 is activated by Epstein–Barr virus LMP1 to mediate EMT and metastasis as an interplay node of multiple pathways in nasopharyngeal carcinoma. Oncogenesis 2017, 6, 402. [Google Scholar] [CrossRef]
- Kim, S.-M.; Oh, S.W.; Park, S.H.; Hur, D.Y.; Hong, S.-W.; Han, S.Y. Epstein-Barr virus-encoded latent membrane protein 1 induces epithelial to mesenchymal transition by inducing V-set Ig domain containing 4 (VSIG4) expression via NF-kB in renal tubular epithelial HK-2 cells. Biochem. Biophys Res. Commun. 2017, 492, 316–322. [Google Scholar] [CrossRef] [PubMed]
- Horikawa, T.; Yang, J.; Kondo, S.; Yoshizaki, T.; Joab, I.; Furukawa, M.; Pagano, J.S. Twist and epithelial-mesenchymal transition are induced by the EBV oncoprotein latent membrane protein 1 and are associated with metastatic nasopharyngeal carcinoma. Cancer Res. 2007, 67, 1970–1978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horikawa, T.; Yoshizaki, T.; Kondo, S.; Furukawa, M.; Kaizaki, Y.; Pagano, J.S. Epstein-Barr Virus latent membrane protein 1 induces Snail and epithelial–mesenchymal transition in metastatic nasopharyngeal carcinoma. Br. J. Cancer. 2011, 104, 1160. [Google Scholar] [CrossRef] [PubMed]
- Wasil, L.; Shair, K. Epstein–Barr virus LMP1 induces focal adhesions and epithelial cell migration through effects on integrin-α5 and N-cadherin. Oncogenesis 2015, 4, e171. [Google Scholar] [CrossRef] [Green Version]
- Lin, Z.; Wan, X.; Jiang, R.; Deng, L.; Gao, Y.; Tang, J.; Yang, Y.; Zhao, W.; Yang, X.; Yao, K.; et al. Epstein-Barr virus-encoded latent membrane protein 2A promotes the epithelial-mesenchymal transition in nasopharyngeal carcinoma via metastatic tumor antigen 1 and mechanistic target of rapamycin signaling induction. J. Virol. 2014, 88, 11872–11885. [Google Scholar] [CrossRef] [Green Version]
- Fotheringham, J.A.; Coalson, N.E.; Raab-Traub, N. Fotheringham JA, Coalson NE, Raab-Traub N. Epstein-Barr virus latent membrane protein-2A induces ITAM/Syk-and Akt-dependent epithelial migration through αv-integrin membrane translocation. J. Virol. 2012, 86, 10308–10320. [Google Scholar] [CrossRef] [Green Version]
- Gaur, N.; Gandhi, J.; Robertson, E.S.; Verma, S.C.; Kaul, R. Epstein–Barr virus latent antigens EBNA3C and EBNA1 modulate epithelial to mesenchymal transition of cancer cells associated with tumor metastasis. Tumour Biol. 2015, 36, 3051–3060. [Google Scholar] [CrossRef]
- Park, G.B.; Kim, D.; Kim, Y.S.; Kim, S.; Lee, H.-K.; Yang, J.W.; Hur, D.Y. The Epstein-Barr Virus Causes Epithelial–Mesenchymal Transition in Human Corneal Epithelial Cells Via Syk/Src and Akt/Erk Signaling Pathways. Invest. Ophthalmol. Vis. Sci. 2014, 55, 1770–1779. [Google Scholar] [CrossRef] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Forward (5′- -3′) | Reverse (5′- -3′) |
|---|---|---|
| qRT-PCR Primers | ||
| BRLF1 | GGCCCAAAAATTGCAGATGT | CCCACGGGCGAGAATG |
| BZLF1 | TCCGACTGGGTCGTGGTT | GCTGCATAAGCTTGATAAGCATTC |
| BMRF1 | GCCGTTGAGGCCCACGTTGT | TGGGAATGGCAGGCGAGGGT |
| BALF4 | AACCTTTGACTCGACCATCG | ACCTGCTCTTCGATGCACTT |
| BLLF1 | GCCGTTGAGGCCCACGTTGT | TGGGAATGGCAGGCGAGGGT |
| EBER1 | CTACGCTGCCCTAGAGGTTTT | CAGCTGGTACTTGACCGAAGA |
| EBER2 | CCTAGTGGTTTCGGACACACC | AATAGCGGACAAGCCGAATAC |
| LMP1 | TCCAGAATTGACGGAAGAGGTT | GCCACCGTCTGTCATCGAA |
| LMP2A | CTCCCTACTCTCCACGGGAT | AGGTAGGGCGCAACAATTACA |
| EBNA1 | GGTCGTGGACGTGGAGAAAA | GGTGGAGACCCGGATGATG |
| ZO-1 | GAAGGAGTTGAGCAGGAAATCTA | AGGACTCAGCAGTGTTTCACC |
| CDH1 | AAGTGCTGCAGCCAAAGACAGA | AAATTGCCAGGCTCAATGACAAG |
| CDH2 | TCAGTGGCGGAGATCCTACT | GTGCTGAATTCCCTTGGCTA |
| VIM | CGCCATCAACACCGAGTTC | ATCTTATTCTGCTGCTCCAGGAA |
| SNAIL1 | CAGGACTCTAATCCAGAGTTTACCT | ACAGAGTCCCAGATGAGCATTG |
| ZEB1 | CATCTTGAGCTGAATTTGGGTAACA | CCTGAAATGACCTGAAGCATGAA |
| KLF4 | CATCTTTCTCCACGTTCGCGT | CGGATCGGATAGGTGAAGCTG |
| GAPDH | AATCCCATCACCATCTTCCA | TGGACTCCACGACGTACTCA |
| β-actin | TGCCGACAGGATGCAGAA | GCCGATCCACACGGAGTACT |
| qPCR Primers | ||
| EBNA1 gene | GGAGCCTGACCTGTGATCGT | TAGGCCATTTCCAGGTCCTGTA |
| LMP1 gene | TCTCCTTTGGCTCCTCCTGT | TCGGTAGCTTGTTGAGGGTG |
| EBV genome | ATGTAAATAAAACCGTGACAGCTCAT | TTACCCAACGGGAAGCATATG |
| β-actin gene | GCCATGGTTGTGCCATTACA | GGCCAGGTTCTCTTTTTATTTCTG |
| Characteristic | FFPE OSCC Tissues N = 165 |
|---|---|
| Gender (%) | |
| Female | 107 (64.8) |
| Male | 58 (35.2) |
| Median age of patients (range) | 70 (20–91) |
| Histology grade of patients (%) | |
| Well differentiated, EBV positive | 49 (29.7) |
| Well differentiated, EBV negative | 53 (32.1) |
| Moderately + poorly differentiated, EBV positive | 19 (11.5) |
| Moderately + poorly differentiated, EBV negative | 44 (26.7) |
| EBV status (%) | |
| EBV positive | 68 (41.2) |
| EBV negative | 97 (58.8) |
| Location of tumor (%) | |
| Buccal mucosa | 65 (39.4) |
| Tongue | 54 (32.7) |
| Lip | 30 (18.2) |
| Other | 16 (9.7) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Heawchaiyaphum, C.; Iizasa, H.; Ekalaksananan, T.; Burassakarn, A.; Kiyono, T.; Kanehiro, Y.; Yoshiyama, H.; Pientong, C. Epstein–Barr Virus Infection of Oral Squamous Cells. Microorganisms 2020, 8, 419. https://doi.org/10.3390/microorganisms8030419
Heawchaiyaphum C, Iizasa H, Ekalaksananan T, Burassakarn A, Kiyono T, Kanehiro Y, Yoshiyama H, Pientong C. Epstein–Barr Virus Infection of Oral Squamous Cells. Microorganisms. 2020; 8(3):419. https://doi.org/10.3390/microorganisms8030419
Chicago/Turabian StyleHeawchaiyaphum, Chukkris, Hisashi Iizasa, Tipaya Ekalaksananan, Ati Burassakarn, Tohru Kiyono, Yuichi Kanehiro, Hironori Yoshiyama, and Chamsai Pientong. 2020. "Epstein–Barr Virus Infection of Oral Squamous Cells" Microorganisms 8, no. 3: 419. https://doi.org/10.3390/microorganisms8030419