Genomic Epidemiology of Carbapenemase Producing Klebsiella pneumoniae Strains at a Northern Portuguese Hospital Enables the Detection of a Misidentified Klebsiella variicola KPC-3 Producing Strain

 , , ,
, , , 
 and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Bacterial Isolates
2.2. Whole-Genome Sequencing
2.3. Phylogenetic Analysis
2.4. In Silico MLST, Plasmid Replicons, Drug Resistance Associated Genes and Capsular Types
3. Results
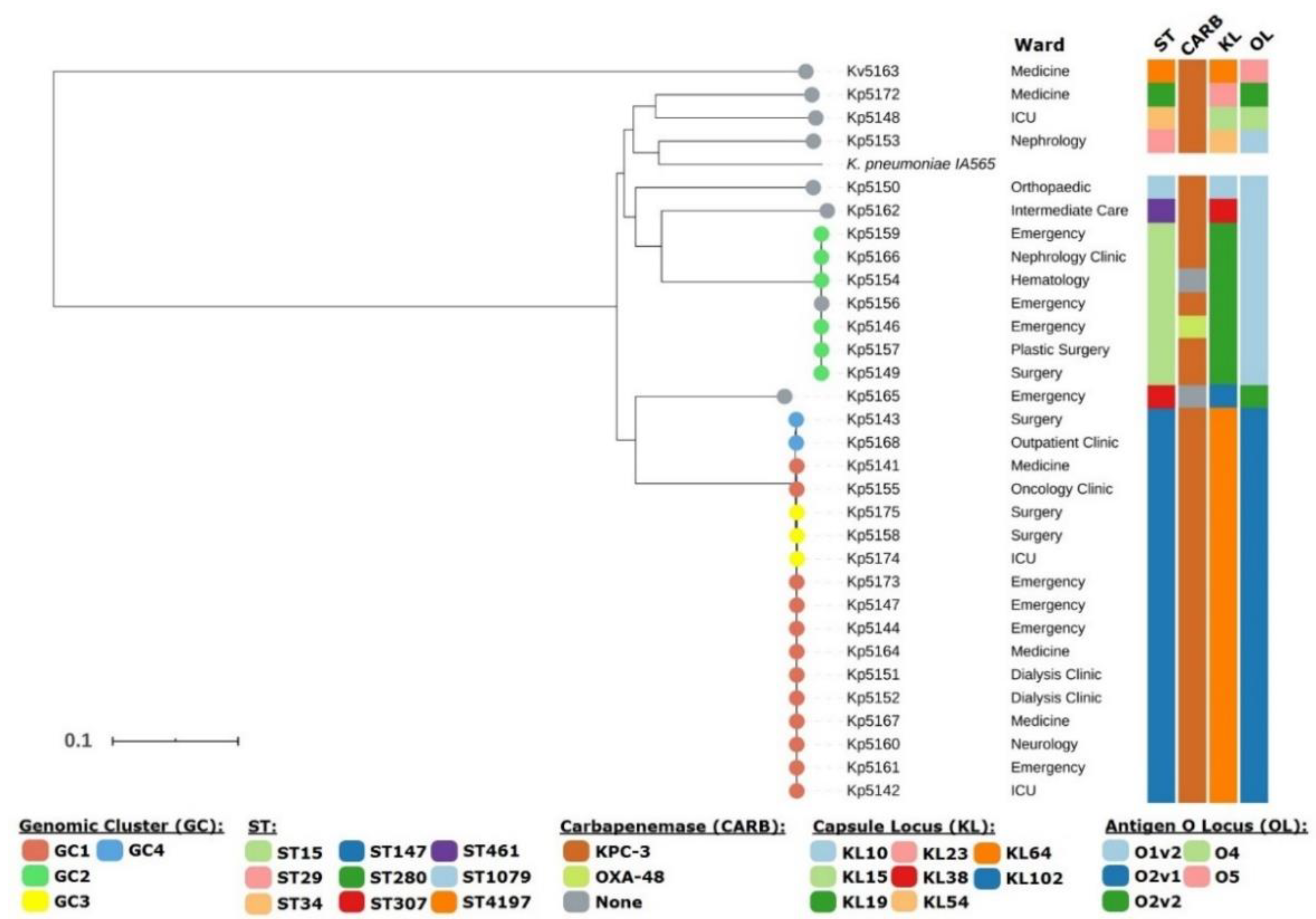
3.1. Whole Genome Sequencing Enables Identification of K. variicola Misidentified as K. pneumoniae
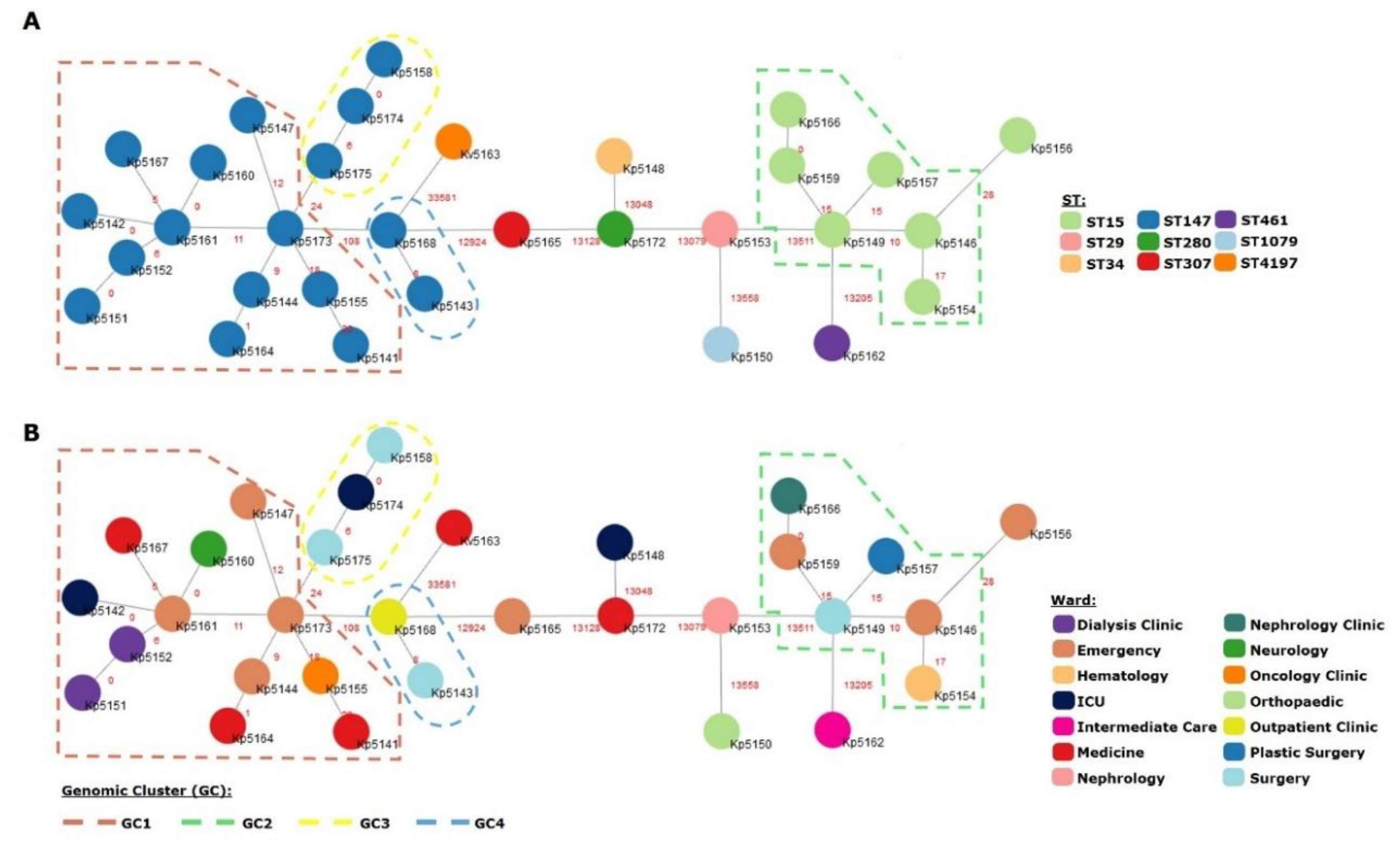
3.2. Genomic Population Structure and High-Resolution Phylogenomic Analysis Unveils Multiple Healthcare-Associated Transmission Clusters
3.3. KPC-3 as the Main Driver of Carbapenem Resistance in Northern Portugal and First Description of KPC-3 Producing K. variicola
3.4. Virulome of K. pneumoniae Highlights the Occurrence of Multiple Virulence Factors and ST-Specific Patterns
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Brisse, S.; Verhoef, J. Phylogenetic diversity of Klebsiella pneumoniae and Klebsiella oxytoca clinical isolates revealed by randomly amplified polymorphic DNA, gyrA and parC genes sequencing and automated ribotyping. Int. J. Syst. Evol. Microbiol. 2001, 51, 915–924. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, C.; Passet, V.; Rakotondrasoa, A.; Diallo, T.A.; Criscuolo, A.; Brisse, S. Description of Klebsiella africanensis sp. nov., Klebsiella variicola subsp. tropicalensis subsp. nov. and Klebsiella variicola subsp. variicola subsp. nov. Res. Microbiol. 2019, 170, 165–170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez-Medina, N.; Barrios-Camacho, H.; Duran-Bedolla, J.; Garza-Ramos, U. Klebsiella variicola: An emerging pathogen in humans. Emerg. Microbes Infect. 2019, 8, 973–988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maatallah, M.; Vading, M.; Kabir, M.H.; Bakhrouf, A.; Kalin, M.; Nauclér, P.; Brisse, S.; Giske, C.G. Klebsiella variicola Is a Frequent Cause of Bloodstream Infection in the Stockholm Area, and Associated with Higher Mortality Compared to K. pneumoniae. PLoS ONE 2014, 9, e113539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garza-Ramos, U.; Moreno-Dominguez, S.; Hernandez-Castro, R.; Silva-Sanchez, J.; Barrios, H.; Reyna-Flores, F.; Sanchez-Perez, A.; Carrillo-Casas, E.M.; Sanchez-Leon, M.C.; Moncada-Barron, D. Identification and Characterization of Imipenem-Resistant Klebsiella pneumoniae and Susceptible Klebsiella variicola Isolates Obtained from the Same Patient. Microb. Drug Resist. 2016, 22, 179–184. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Feng, Y.; Tang, G.; Lin, J.; Huang, W.; Qiao, F.; Zong, Z. Carbapenem-resistant Isolates of the Klebsiella pneumoniae Complex in Western China: The Common ST11 and the Surprising Hospital-specific Types. Clin. Infect. Dis. 2018, 67, S263–S265. [Google Scholar] [CrossRef]
- Hopkins, K.L.; Findlay, J.; Doumith, M.; Mather, B.; Meunier, D.; D’Arcy, S.; Pike, R.; Mustafa, N.; Howe, R.; Wootton, M.; et al. IMI-2 carbapenemase in a clinical Klebsiella variicola isolated in the UK. J. Antimicrob. Chemother. 2017, 72, 2129–2131. [Google Scholar] [CrossRef]
- Rosenblueth, M.; Martinez, L.; Silva, J.; Martinez-Romero, E. Klebsiella variicola, a novel species with clinical and plant-associated isolates. Syst. Appl. Microbiol. 2004, 27, 27–35. [Google Scholar] [CrossRef] [Green Version]
- Potter, R.F.; Lainhart, W.; Twentyman, J.; Wallace, M.A.; Wang, B.; Burnham, C.A.; Rosen, D.A.; Dantas, G. Population Structure, Antibiotic Resistance, and Uropathogenicity of Klebsiella variicola. mBio 2018, 9. [Google Scholar] [CrossRef] [Green Version]
- Andrade, B.G.; de Veiga Ramos, N.; Marin, M.F.; Fonseca, E.L.; Vicente, A.C. The genome of a clinical Klebsiella variicola strain reveals virulence-associated traits and a pl9-like plasmid. FEMS Microbiol. Lett. 2014, 360, 13–16. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.; Zhai, Y.; Zhang, Z.; Li, D.; Wang, Z.; Li, J.; He, Z.; Hu, S.; Kang, Y.; Gao, Z. Complete Genomic Analysis of a Kingdom-Crossing Klebsiella variicola Isolate. Front. Microbiol. 2018, 9, 2428. [Google Scholar] [CrossRef] [PubMed]
- The European Committee on Antimicrobial Susceptibility Testing. Breakpoint Tables for Interpretation of MICs and Zone Diameters—Version 8.0: European Society of Clinical Microbiology and Infectious Diseases; EUCAST: Växjö, Sweden, 2018. [Google Scholar]
- Tsakris, A.; Kristo, I.; Poulou, A.; Themeli-Digalaki, K.; Ikonomidis, A.; Petropoulou, D.; Pournaras, S.; Sofianou, D. Evaluation of boronic acid disk tests for differentiating KPC-possessing Klebsiella pneumoniae isolates in the clinical laboratory. J. Clin. Microbiol. 2009, 47, 362–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [Green Version]
- Coll, F.; Phelan, J.; Hill-Cawthorne, G.A.; Nair, M.B.; Mallard, K.; Ali, S.; Abdallah, A.M.; Alghamdi, S.; Alsomali, M.; Ahmed, A.O.; et al. Genome-wide analysis of multi- and extensively drug-resistant Mycobacterium tuberculosis. Nat. Genet. 2018, 50, 307–316. [Google Scholar] [CrossRef] [Green Version]
- Anisimova, M.; Gascuel, O. Approximate likelihood-ratio test for branches: A fast, accurate, and powerful alternative. Syst. Biol. 2006, 55, 539–552. [Google Scholar] [CrossRef]
- Schliep, K.P. Phangorn: Phylogenetic analysis in R. Bioinformatics 2011, 27, 592–593. [Google Scholar] [CrossRef] [Green Version]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v4: Recent updates and new developments. Nucleic Acids Res. 2019, 47, W256–W259. [Google Scholar] [CrossRef] [Green Version]
- Nascimento, M.; Sousa, A.; Ramirez, M.; Francisco, A.P.; Carrico, J.A.; Vaz, C. PHYLOViZ 2.0: Providing scalable data integration and visualization for multiple phylogenetic inference methods. Bioinformatics 2017, 33, 128–129. [Google Scholar] [CrossRef] [PubMed]
- Sherry, N.L.; Lane, C.R.; Kwong, J.C.; Schultz, M.; Sait, M.; Stevens, K.; Ballard, S.; Goncalves da Silva, A.; Seemann, T.; Gorrie, C.L.; et al. Genomics for Molecular Epidemiology and Detecting Transmission of Carbapenemase-Producing Enterobacterales in Victoria, Australia, 2012 to 2016. J. Clin. Microbiol. 2019, 57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carattoli, A.; Zankari, E.; Garcia-Fernandez, A.; Voldby Larsen, M.; Lund, O.; Villa, L.; Moller Aarestrup, F.; Hasman, H. In silico detection and typing of plasmids using PlasmidFinder and plasmid multilocus sequence typing. Antimicrob. Agents Chemother. 2014, 58, 3895–3903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katoh, K.; Rozewicki, J.; Yamada, K.D. MAFFT online service: Multiple sequence alignment, interactive sequence choice and visualization. Brief Bioinform. 2019, 20, 1160–1166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larsen, M.V.; Cosentino, S.; Lukjancenko, O.; Saputra, D.; Rasmussen, S.; Hasman, H.; Sicheritz-Ponten, T.; Aarestrup, F.M.; Ussery, D.W.; Lund, O. Benchmarking of methods for genomic taxonomy. J. Clin. Microbiol. 2014, 52, 1529–1539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perdigao, J.; Modesto, A.; Pereira, A.L.; Neto, O.; Matos, V.; Godinho, A.; Phelan, J.; Charleston, J.; Spadar, A.; de Sessions, P.F.; et al. Whole-genome sequencing resolves a polyclonal outbreak by extended-spectrum beta-lactam and carbapenem-resistant Klebsiella pneumoniae in a Portuguese tertiary-care hospital. Microb. Genom. 2020. [CrossRef]
- Poirel, L.; Bonnin, R.A.; Nordmann, P. Genetic features of the widespread plasmid coding for the carbapenemase OXA-48. Antimicrob. Agents Chemother. 2012, 56, 559–562. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Ye, M.; Li, X.; Li, J.; Deng, Z.; Yao, Y.F.; Ou, H.Y. Identification and Characterization of an Antibacterial Type VI Secretion System in the Carbapenem-Resistant Strain Klebsiella pneumoniae HS11286. Front. Cell Infect. Microbiol. 2017, 7, 442. [Google Scholar] [CrossRef] [Green Version]
- Hsieh, P.F.; Lu, Y.R.; Lin, T.L.; Lai, L.Y.; Wang, J.T. Klebsiella pneumoniae Type VI Secretion System Contributes to Bacterial Competition, Cell Invasion, Type-1 Fimbriae Expression, and In Vivo Colonization. J. Infect. Dis. 2019, 219, 637–647. [Google Scholar] [CrossRef] [Green Version]
- Fonseca, E.L.; Ramos, N.D.; Andrade, B.G.; Morais, L.L.; Marin, M.F.; Vicente, A.C. A one-step multiplex PCR to identify Klebsiella pneumoniae, Klebsiella variicola, and Klebsiella quasipneumoniae in the clinical routine. Diagn. Microbiol. Infect. Dis. 2017, 87, 315–317. [Google Scholar] [CrossRef]
- Huynh, B.T.; Passet, V.; Rakotondrasoa, A.; Diallo, T.; Kerleguer, A.; Hennart, M.; Lauzanne, A.; Herindrainy, P.; Seck, A.; Bercion, R.; et al. Klebsiella pneumoniae carriage in low-income countries: Antimicrobial resistance, genomic diversity and risk factors. Gut. Microbes 2020, 11, 1287–1299. [Google Scholar] [CrossRef] [PubMed]
- Perez, F.; Rudin, S.D.; Marshall, S.H.; Coakley, P.; Chen, L.; Kreiswirth, B.N.; Rather, P.N.; Hujer, A.M.; Toltzis, P.; van Duin, D.; et al. OqxAB, a quinolone and olaquindox efflux pump, is widely distributed among multidrug-resistant Klebsiella pneumoniae isolates of human origin. Antimicrob. Agents Chemother. 2013, 57, 4602–4603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodrigues, C.; Bavlovic, J.; Machado, E.; Amorim, J.; Peixe, L.; Novais, A. KPC-3-Producing Klebsiella pneumoniae in Portugal Linked to Previously Circulating Non-CG258 Lineages and Uncommon Genetic Platforms (Tn4401d-IncFIA and Tn4401d-IncN). Front. Microbiol. 2016, 7, 1000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caneiras, C.; Calisto, F.; Jorge da Silva, G.; Lito, L.; Melo-Cristino, J.; Duarte, A. First Description of Colistin and Tigecycline-Resistant Acinetobacter baumannii Producing KPC-3 Carbapenemase in Portugal. Antibiotics 2018, 7, 96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caneiras, C.; Lito, L.; Mayoralas-Alises, S.; Diaz-Lobato, S.; Melo-Cristino, J.; Duarte, A. Virulence and resistance determinants of Klebsiella pneumoniae isolated from a Portuguese tertiary university hospital centre over a 31-year period. Enferm. Infecc. Microbiol. Clin. 2019, 37, 387–393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonnin, R.A.; Jousset, A.B.; Chiarelli, A.; Emeraud, C.; Glaser, P.; Naas, T.; Dortet, L. Emergence of New Non-Clonal Group 258 High-Risk Clones among Klebsiella pneumoniae Carbapenemase-Producing K. pneumoniae Isolates, France. Emerg. Infect. Dis. 2020, 26, 1212–1220. [Google Scholar] [CrossRef] [PubMed]
- Wyres, K.L.; Lam, M.M.C.; Holt, K.E. Population genomics of Klebsiella pneumoniae. Nat. Rev. Microbiol. 2020, 18, 344–359. [Google Scholar] [CrossRef]
- Enne, V.I.; Livermore, D.M.; Stephens, P.; Hall, L.M. Persistence of sulphonamide resistance in Escherichia coli in the UK despite national prescribing restriction. Lancet 2001, 357, 1325–1328. [Google Scholar] [CrossRef]
- Paczosa, M.K.; Mecsas, J. Klebsiella pneumoniae: Going on the Offense with a Strong Defense. Microbiol. Mol. Biol. Rev. 2016, 80, 629–661. [Google Scholar] [CrossRef] [Green Version]
- Hsieh, P.F.; Wu, M.C.; Yang, F.L.; Chen, C.T.; Lou, T.C.; Chen, Y.Y.; Wu, S.H.; Sheu, J.C.; Wang, J.T. D-galactan II is an immunodominant antigen in O1 lipopolysaccharide and affects virulence in Klebsiella pneumoniae: Implication in vaccine design. Front. Microbiol. 2014, 5, 608. [Google Scholar] [CrossRef]
- Di Martino, P.; Cafferini, N.; Joly, B.; Darfeuille-Michaud, A. Klebsiella pneumoniae type 3 pili facilitate adherence and biofilm formation on abiotic surfaces. Res. Microbiol. 2003, 154, 9–16. [Google Scholar] [CrossRef]
- Chen, Y.T.; Chang, H.Y.; Lai, Y.C.; Pan, C.C.; Tsai, S.F.; Peng, H.L. Sequencing and analysis of the large virulence plasmid pLVPK of Klebsiella pneumoniae CG43. Gene 2004, 337, 189–198. [Google Scholar] [CrossRef] [PubMed]
- Lam, M.M.; Wyres, K.L.; Judd, L.M.; Wick, R.R.; Jenney, A.; Brisse, S.; Holt, K.E. Tracking key virulence loci encoding aerobactin and salmochelin siderophore synthesis in Klebsiella pneumoniae. Genome Med. 2018, 10, 77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sorsa, L.J.; Dufke, S.; Heesemann, J.; Schubert, S. Characterization of an iroBCDEN gene cluster on a transmissible plasmid of uropathogenic Escherichia coli: Evidence for horizontal transfer of a chromosomal virulence factor. Infect. Immun. 2003, 71, 3285–3293. [Google Scholar] [CrossRef] [Green Version]
- Ma, L.C.; Fang, C.T.; Lee, C.Z.; Shun, C.T.; Wang, J.T. Genomic heterogeneity in Klebsiella pneumoniae strains is associated with primary pyogenic liver abscess and metastatic infection. J. Infect. Dis. 2005, 192, 117–128. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
| ID | Isolates | Patient | Sample | Hospital Ward | Date | ST |
|---|---|---|---|---|---|---|
| 5141 | K. pneumoniae | P1 | Rectal swab | Medicine | 2 May 2018 | ST147 |
| 5142 | K. pneumoniae | P2 | Rectal swab | ICU | 3 May 2018 | ST147 |
| 5144 | K. pneumoniae | P3 | Rectal swab | Emergency | 4 May 2018 | ST147 |
| 5147 | K. pneumoniae | P4 | Urine | Emergency | 6 May 2018 | ST147 |
| 5151 | K. pneumoniae | P5 | Dialysis liquid | Dialysis Clinic | 15 May 2018 | ST147 |
| 5152 | K. pneumoniae | Purulent exudate | Dialysis Clinic | 15 May 2018 | ST147 | |
| 5143 | K. pneumoniae | P6 | Rectal swab | Surgery | 16 May 2018 | ST147 |
| 5160 | K. pneumoniae | P7 | Rectal swab | Neurology | 25 May 2018 | ST147 |
| 5161 | K. pneumoniae | P8 | Rectal swab | Emergency | 26 May 2018 | ST147 |
| 5155 | K. pneumoniae | P9 | Urine | Oncology Clinic | 29 May 2018 | ST147 |
| 5158 | K. pneumoniae | P10 | Urine | Surgery | 4 June 2018 | ST147 |
| 5164 | K. pneumoniae | P11 | Urine | Medicine | 4 June 2018 | ST147 |
| 5167 | K. pneumoniae | P12 | Rectal swab | Medicine | 4 June 2018 | ST147 |
| 5168 | K. pneumoniae | P13 | Urine | Outpatient Clinic | 11 June 2018 | ST147 |
| 5173 | K. pneumoniae | P14 | Urine | Emergency | 17 June 2018 | ST147 |
| 5174 | K. pneumoniae | P15 | Blood | Emergency | 19 June 2018 | ST147 |
| 5175 | K. pneumoniae | P16 | Rectal swab | Surgery | 19 June 2018 | ST147 |
| 5146 | K. pneumoniae | P17 | Urine | Emergency | 3 May 2018 | ST15 |
| 5149 | K. pneumoniae | P18 | Hepatic liquid | Surgery | 9 May 2018 | ST15 |
| 5154 | K. pneumoniae | P19 | Blood | Hematology | 28 May 2018 | ST15 |
| 5159 | K. pneumoniae | P20 | Rectal swab | Emergency | 25 May 2018 | ST15 |
| 5156 | K. pneumoniae | Sputum | Emergency | 30 May 2018 | ST15 | |
| 5157 | K. pneumoniae | P21 | Biopsy | Plastic Surgery | 4 June 2018 | ST15 |
| 5166 | K. pneumoniae | P22 | Urina | Nephrology clinic | 6 June 2018 | ST15 |
| 5148 | K. pneumoniae | P23 | Urine | ICU | 7 May 2018 | ST34 |
| 5150 | K. pneumoniae | P24 | Urine | Orthopaedic | 13 May 2018 | ST1079 |
| 5153 | K. pneumoniae | P25 | RTS/DLC a | Nephrology | 21 May 2018 | ST29 |
| 5162 | K. pneumoniae | P26 | Rectal swab | ICU | 29 May 2018 | ST461 |
| 5165 | K. pneumoniae | P27 | Rectal swab | Emergency | 5 June 2018 | ST307 |
| 5163 | K. variicola | P28 | Rectal swab | Medicine | 29 May 2018 | ST4197 |
| 5172 | K. pneumoniae | Rectal swab | Medicine | 18 June 2018 | ST280 |
| Isolates ID | Sequence Type | Genomic Cluster | bla_Carb | bla_ESBL | bla_narrow Spectrum | Aminoglycoside | aac(6′)-Ib-cr5 | qnr | Phe | Rif | Fos | oqxAB | tmp | qacEdelta1 | sul | tet | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Kp5141 | ST147 | GC1 | KPC-3 | CTX-M-15 | SHV-11 | TEM-1 | OXA-1 | aph(6)-Id; aph(3″)-Ib | aac(3)-IIa | aac(6′)-Ib-cr5 | catB3 | fosA | oqxAB | dfrA14 | sul2 | ||||
| Kp5142 | ST147 | GC1 | KPC-3 | SHV-11 | aadA1 | fosA | oqxAB | dfrA1 | qacEdelta1 | sul1 | |||||||||
| Kp5144 | ST147 | GC1 | KPC-3 | SHV-11 | aadA16 | aac(6′)-Ib-cr5 | qnrB6 | - | arr-3 | fosA | oqxAB | dfrA27 | sul1 | ||||||
| Kp5147 | ST147 | GC1 | KPC-3 | - | SHV-11 | aadA16 | aac(6′)-Ib-cr5 | - | arr-3 | fosA | oqxAB | dfrA27 | qacEdelta1 | sul1 | |||||
| Kp 5151 | ST147 | GC1 | KPC-3 | - | SHV-11 | aadA16 | aac(6′)-Ib-cr5 | qnrB6 | - | arr-3 | fosA | oqxAB | dfrA27 | qacEdelta1 | sul1 | ||||
| Kp5152 | ST147 | GC1 | KPC-3 | - | SHV-11 | aadA16 | aac(6′)-Ib-cr5 | qnrB6 | - | arr-3 | fosA | oqxAB | dfrA27 | qacEdelta1 | sul1 | ||||
| Kp5143 | ST147 | GC4 | KPC-3 | SHV-12 | TEM-1 | aph(6)-Id; aph(3″)-Ib | qnrB1 | fosA | oqxAB | dfrA14 | qacEdelta1 | sul2 | tet(A) | ||||||
| Kp5160 | ST147 | GC1 | KPC-3 | - | SHV-11 | aadA1 | - | - | fosA | oqxAB | dfrA1 | qacEdelta1 | sul1 | ||||||
| Kp5161 | ST147 | GC1 | KPC-3 | - | SHV-11 | aadA1 | - | - | fosA | oqxAB | dfrA1 | qacEdelta1 | sul1 | ||||||
| Kp5155 | ST147 | GC1 | KPC-3 | SHV-11 | TEM-33 | - | - | - | fosA | oqxAB | - | ||||||||
| Kp5158 | ST147 | GC1 | KPC-3 | - | SHV-1 | aadA16 | aac(6′)-Ib-cr5 | qnrB6 | - | arr-3 | fosA | oqxAB | dfrA27 | qacEdelta1 | sul1 | ||||
| Kp5164 | ST147 | GC1 | KPC-3 | - | SHV-11 | aadA16 | aac(6′)-Ib-cr5 | qnrB6 | - | arr-3 | fosA | oqxAB | dfrA27 | qacEdelta1 | sul1 | ||||
| Kp5167 | ST147 | GC1 | KPC-3 | - | SHV-11 | aadA1 | - | - | fosA | oqxAB | dfrA1 | qacEdelta1 | sul1 | ||||||
| Kp5168 | ST147 | GC1 | KPC-3 | SHV-12 | TEM-1 | OXA-9 | aph(6)-Id; aph(3″)-Ib | aac(6′)-Ib; aadA1 | qnrB1 | - | - | fosA | oqxAB | dfrA14 | sul2 | tet(A) | |||
| Kp5173 | ST147 | GC1 | KPC-3 | - | SHV-11 | aadA16 | aac(6′)-Ib-cr5 | qnrB6 | - | arr-3 | fosA | oqxAB | dfrA27 | qacEdelta1 | sul1 | ||||
| Kp5174 | ST147 | GC3 | KPC-3 | - | SHV-1 | aadA16 | aac(6′)-Ib-cr5 | qnrB6 | - | arr-3 | fosA | oqxAB | dfrA27 | qacEdelta1 | sul1 | ||||
| Kp5175 | ST147 | GC3 | KPC-3 | - | SHV-11 | aadA16 | aac(6′)-Ib-cr5 | qnrB6 | - | arr-3 | fosA | oqxAB | dfrA27 | qacEdelta1 | sul1 | ||||
| Kp5146 | ST15 | GC2 | OXA-48 | CTX-M-15 | SHV-28 | TEM-1 | OXA-1 | aph(6)-Id; aph(3″)-Ib | aac(3)-IIa | aac(6′)-Ib-cr5 | qnrB1 | catA1; catB3 | - | fosA | oqxAB | dfrA14 | sul2 | ||
| Kp5149 | ST15 | GC2 | KPC-3 | - | SHV-28 | TEM-1 | OXA-9 | aph(6)-Id; aph(3″)-Ib | aac(6′)-Ib; aadA1 | - | - | fosA | oqxAB | dfrA14 | sul2 | ||||
| Kp5154 | ST15 | GC2 | CTX-M-15 | SHV-28 | TEM-1 | aph(6)-Id; aph(3″)-Ib | aac(3)-IIa | qnrB1 | catA1; catB3 | - | fosA | oqxAB | dfrA14 | sul2 | |||||
| Kp5159 | ST15 | GC2 | KPC-3 | - | SHV-28 | TEM-1 | OXA-9 | aph(6)-Id; aph(3″)-Ib | aadA1 | qnrB1 | catA1 | - | fosA | oqxAB | dfrA14 | sul2 | tet(A) | ||
| Kp5156 | ST15 | KPC-3 | CTX-M-15 | TEM-1 | OXA-9 | aph(6)-Id; aph(3″)-Ib | aac(6′)-Ib; aadA1 | qnrB1 | - | - | fosA | oqxAB | dfrA14 | sul2 | |||||
| Kp5157 | ST15 | GC2 | KPC-3 | - | SHV-28 | TEM-1 | OXA-9 | aph(6)-Id; aph(3″)-Ib | aac(6′)-Ib; aadA1 | - | - | fosA | oqxAB | dfrA14 | sul2 | ||||
| Kp5166 | ST15 | GC2 | KPC-3 | - | SHV-28 | TEM-1 | OXA-9 | aph(6)-Id; aph(3″)-Ib | aadA1 | qnrB1 | catA1 | - | fosA | oqxAB | dfrA14 | sul2 | tet(A) | ||
| Kp5148 | ST34 | KPC-3 | - | SHV-26 | - | - | - | fosA | oqxA10; oqxB19 | - | |||||||||
| Kp5150 | ST1079 | KPC-3 | - | SHV-1 | TEM-1 | OXA-9 | aph(6)-Id; aph(3″)-Ib | aac(6′)-Ib; aadA1 | - | - | fosA | oqxA; oqxB19 | dfrA14 | sul2 | |||||
| Kp5153 | ST29 | KPC-3 | - | SHV-187 | TEM-1 | OXA-9 | aph(6)-Id; aph(3″)-Ib | aac(6′)-Ib; aadA1 | qnrS1 | - | - | fosA | oqxA; oqxB25 | dfrA14 | sul2 | ||||
| Kp5162 | ST451 | KPC-3 | - | SHV-1 | - | - | - | fosA | oqxA10; oqxB25 | - | |||||||||
| Kp5165 | ST307 | CTX-M-15 | SHV-28 | TEM-1 | aph(6)-Id; aph(3″)-Ib | aac(3)-IIa; aadA16 | aac(6′)-Ib-cr5 | qnrB6 | catA2 | arr-3 | fosA | oqxA; oqxB19 | dfrA27 | qacEdelta1 | sul1;sul2 | tet(D) | |||
| Kv5163 | ST4197 | KPC-3 | - | LEN-2 | - | - | - | fosA | oqxA; oqxB15 | - | |||||||||
| Kp5172 | ST280 | KPC-3 | CTX-M-15 | SHV-27 | TEM-1 | OXA-1; OXA-9 | aph(6)-Id; aph(3″)-Ib | aac(3)-IIa; aac(6′)-Ib; aadA1 | aac(6′)-Ib-cr5 | qnrB1 | catB3 | - | fosA | oqxA; oqxB19 | dfrA14 | sul2 | tet(A) | ||
| K. pneumoniae | K. variicola | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Replicon | ST147 ** | ST15 | ST461 | ST1079 | ST29 | ST307 | ST280 | ST34 | ST4197 | Identity/% | N° Isolates/% |
| IncFIA (HI1) | 6 | 7 | 1 | 1 | 1 | 1 | 1 | 94.59–100 | 18/58.1 | ||
| IncFIB (pQil)_1_pQil | 1 * | 1 * | 100 | 02/06.4 | |||||||
| IncFIB (K)_1_Kpn3 | 12 (9 *) | 7 * | 1 | 1 | 1 * | 1 | 1 * | 98.93–100 | 24/77.4 | ||
| IncFIB (pKPHS1) | 14 | 97.32–100 | 14/45.5 | ||||||||
| IncFIB (Mar)_1_pNDM | 1 | 1 | 1 * | 99.77–100 | 03/09.7 | ||||||
| IncFII_1_pKP91 | 10 | 5 | 1 | 1 | 1 | 85.47–88.55 | 18/58.1 | ||||
| IncFII (Yp) | 6 | 5 | 1 | 1 | 1 | 85.71 | 14/45.5 | ||||
| IncFII (pMET)_1_pMET1 | 1 | 98.09 | 01/03,2 | ||||||||
| IncR_1 | 4 (2 *) | 2 * | 1 | 99.2–100 | 07/22.5 | ||||||
| IncN_1 | 16 | 1 | 99.42 | 17/54.8 | |||||||
| IncL/M (pOXA-48) | 1 * | 100 | 01/03.2 | ||||||||
| IncX4_1 | 1 | 99.73 | 01/03.2 | ||||||||
| repA-pKPC-2 | 1 | 99.71 | 01/03.2 | ||||||||
| Col (MG828)1 | 1 | 93.51 | 01/03.2 | ||||||||
| ColRNAI_1 | 4 (2 *) | 6 | 1 | 1 | 1 | 1 | 85.5–100 | 14/45.5 | |||
| ColpVC_1 | 1 | 98.45 | 01/03.2 | ||||||||
| Isolates ID | Sequence Type | wzi | K_locus | O_locus | Enterobactin * | Ferric Iron ** | Ybt *** | Type I Fimbriae | Type 3 Fimbriae | Type VI SSE |
|---|---|---|---|---|---|---|---|---|---|---|
| Kp5141 | ST147 | wzi64 | KL64 | O2v1 | entB | ybtQ | fimH | mrkD_12 | tle1; tli1 | |
| Kp5142 | ST147 | wzi64 | KL64 | O2v1 | entB | ybtQ | fimH | mrkD_12 | tle1; tli1 | |
| Kp5144 | ST147 | wzi64 | KL64 | O2v1 | entB | ybtQ | fimH | mrkD_12 | tle1; tli1 | |
| Kp5147 | ST147 | wzi64 | KL64 | O2v1 | entB | ybtQ | fimH | mrkD_12 | tle1; tli1 | |
| Kp5151 | ST147 | wzi64 | KL64 | O2v1 | entB | ybtQ | fimH | mrkD_12 | tle1; tli1 | |
| Kp5152 | ST147 | wzi64 | KL64 | O2v1 | entB | ybtQ | fimH | mrkD_12 | tle1; tli1 | |
| Kp5143 | ST147 | wzi64 | KL64 | O2v1 | entB | |||||
| Kp5160 | ST147 | wzi64 | KL64 | O2v1 | entB | ybtQ | fimH | mrkD_12 | tle1; tli1 | |
| Kp5161 | ST147 | wzi64 | KL64 | O2v1 | entB | ybtQ | fimH | mrkD_12 | tle1; tli1 | |
| Kp5155 | ST147 | wzi64 | KL64 | O2v1 | entB | |||||
| Kp5158 | ST147 | wzi64 | KL64 | O2v1 | entB | ybtQ | fimH | mrkD_12 | tle1; tli1 | |
| Kp5164 | ST147 | wzi64 | KL64 | O2v1 | entB | ybtQ | fimH | mrkD_12 | tle1; tli1 | |
| Kp5167 | ST147 | wzi64 | KL64 | O2v1 | entB | ybtQ | fimH | mrkD_12 | tle1; tli1 | |
| Kp5168 | ST147 | wzi64 | KL64 | O2v1 | entB | fimH | mrkD_12 | tle1; tli1 | ||
| Kp5173 | ST147 | wzi64 | KL64 | O2v1 | entB | ybtQ | fimH | mrkD_12 | tle1; tli1 | |
| Kp5174 | ST147 | wzi64 | KL64 | O2v1 | entB | ybtQ | fimH | mrkD_12 | tle1; tli1 | |
| Kp5175 | ST147 | wzi64 | KL64 | O2v1 | entB | ybtQ | fimH | mrkD_12 | tle1; tli1 | |
| Kp5146 | ST15 | wzi19 | KL19 | O1v2 | entB | kfuA_3 | ybtQ | fimH | ||
| Kp5149 | ST15 | wzi19 | KL19 | O1v2 | entB | kfuA_3 | ybtQ | fimH | ||
| Kp5154 | ST15 | wzi19 | KL19 | O1v2 | entB | kfuA_3 | ybtQ | fimH | ||
| Kp5159 | ST15 | wzi19 | KL19 | O1v2 | entB | kfuA_3 | ybtQ | fimH | ||
| Kp5156 | ST15 | wzi19 | KL19 | O1v2 | entB | kfuA_3 | fimH | |||
| Kp5157 | ST15 | wzi19 | KL19 | O1v2 | entB | kfuA_3 | fimH | |||
| Kp5166 | ST15 | wzi19 | KL19 | O1v2 | entB | kfuA_3 | ybtQ | fimH | ||
| Kp5148 | ST34 | wzi50 | KL15 | O4 | entB | fimH | tle1; tli1 | |||
| Kp5150 | ST1079 | wzi236 | KL10 | O1v2 | entB | fimH | ||||
| Kp5153 | ST29 | wzi115 | KL54 | O1v2 | entB | fimH | ||||
| Kp5162 | ST451 | wzi38 | KL38 | O1v2 | entB | kfuA_26 | fimH | |||
| Kp5165 | ST307 | wzi173 | KL102 | O2v2 | entB | fimH | tle1; tli1 | |||
| Kv5163 | ST4197 | - | KL64 | O5 | entB | kfuA_8 | fimH | tle1; tli1 | ||
| Kp5172 | ST280 | wzi82 | KL23 | O2v2 | entB | fimH | tle1; tli1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Perdigão, J.; Caneiras, C.; Elias, R.; Modesto, A.; Spadar, A.; Phelan, J.; Campino, S.; Clark, T.G.; Costa, E.; Saavedra, M.J.; et al. Genomic Epidemiology of Carbapenemase Producing Klebsiella pneumoniae Strains at a Northern Portuguese Hospital Enables the Detection of a Misidentified Klebsiella variicola KPC-3 Producing Strain. Microorganisms 2020, 8, 1986. https://doi.org/10.3390/microorganisms8121986
Perdigão J, Caneiras C, Elias R, Modesto A, Spadar A, Phelan J, Campino S, Clark TG, Costa E, Saavedra MJ, et al. Genomic Epidemiology of Carbapenemase Producing Klebsiella pneumoniae Strains at a Northern Portuguese Hospital Enables the Detection of a Misidentified Klebsiella variicola KPC-3 Producing Strain. Microorganisms. 2020; 8(12):1986. https://doi.org/10.3390/microorganisms8121986
Chicago/Turabian StylePerdigão, João, Cátia Caneiras, Rita Elias, Ana Modesto, Anton Spadar, Jody Phelan, Susana Campino, Taane G. Clark, Eliana Costa, Maria José Saavedra, and et al. 2020. "Genomic Epidemiology of Carbapenemase Producing Klebsiella pneumoniae Strains at a Northern Portuguese Hospital Enables the Detection of a Misidentified Klebsiella variicola KPC-3 Producing Strain" Microorganisms 8, no. 12: 1986. https://doi.org/10.3390/microorganisms8121986