Inter-Versus Intra-Host Sequence Diversity of pH1N1 and Associated Clinical Outcomes


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Material and Methods
2.1. Sample Collection and Atudy Population
2.2. Ethics Statement
2.3. RNA Extraction and RT-PCR
2.4. Library Preparation and Sequencing
2.5. Data Filtering and Read Processing
2.6. Variant Analysis
2.7. Mutation Rate and Phylogeny Construction
2.8. Analysis of Low-Frequency Variants in the Shadow of Global Variation
2.9. Haplotype Reconstruction and Diversity Analysis
2.10. Statistical Analysis
3. Results
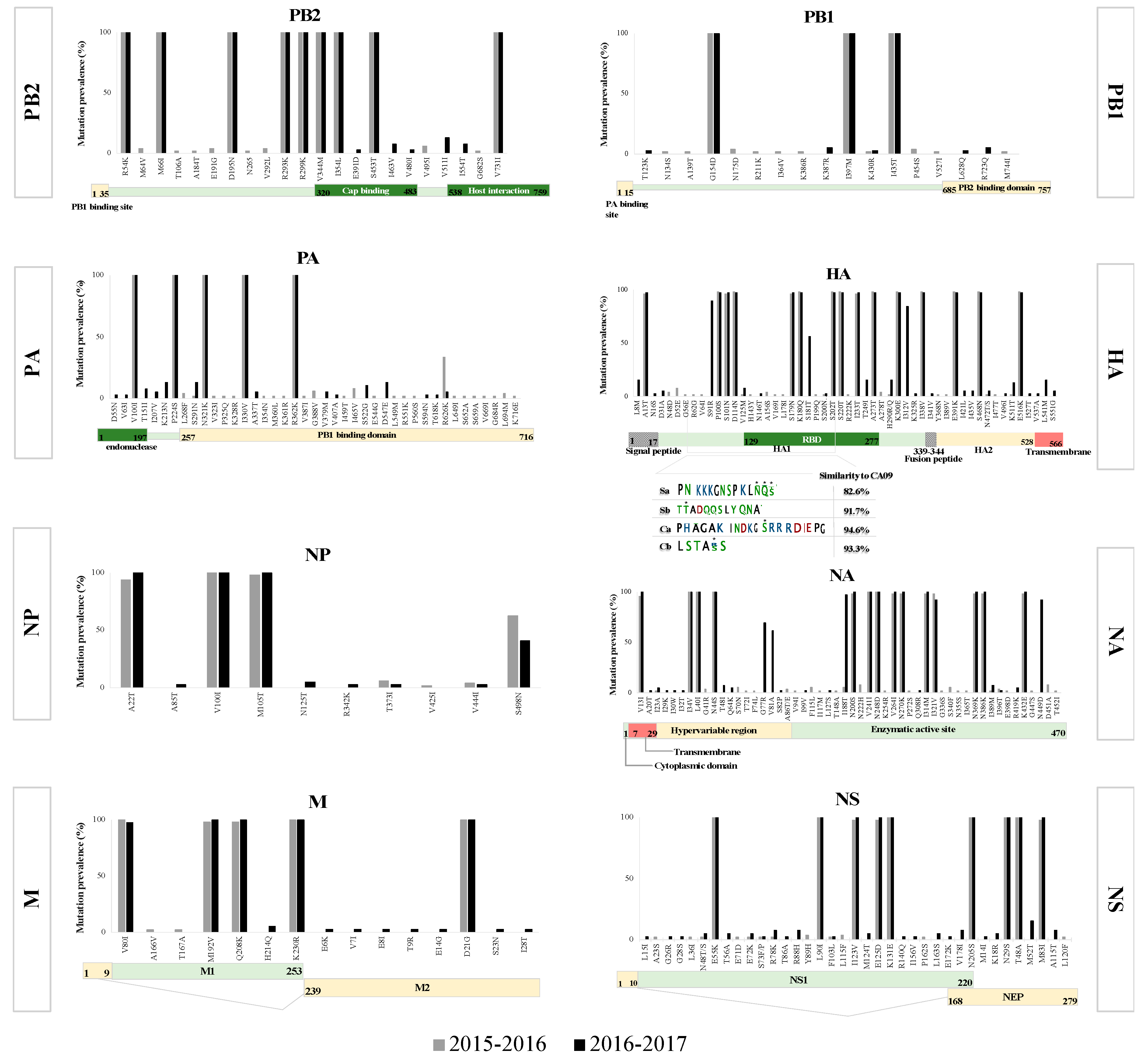
3.1. Molecular Characterization of pH1N1 Genes during the 2015–2017 Period
Phylogenetic Analysis, Mutation Rate, and Selection Pressure
3.2. Mutation Analysis
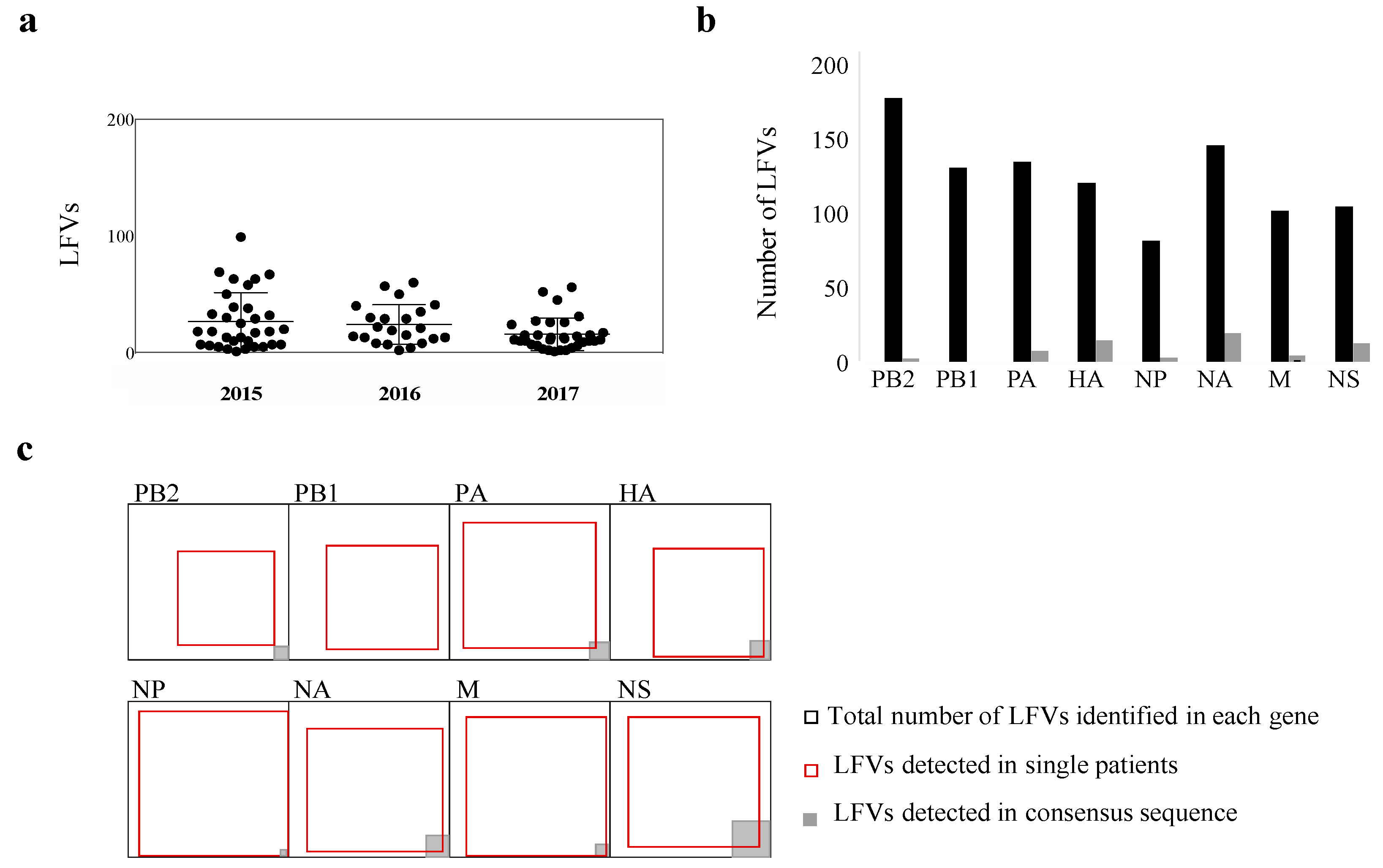
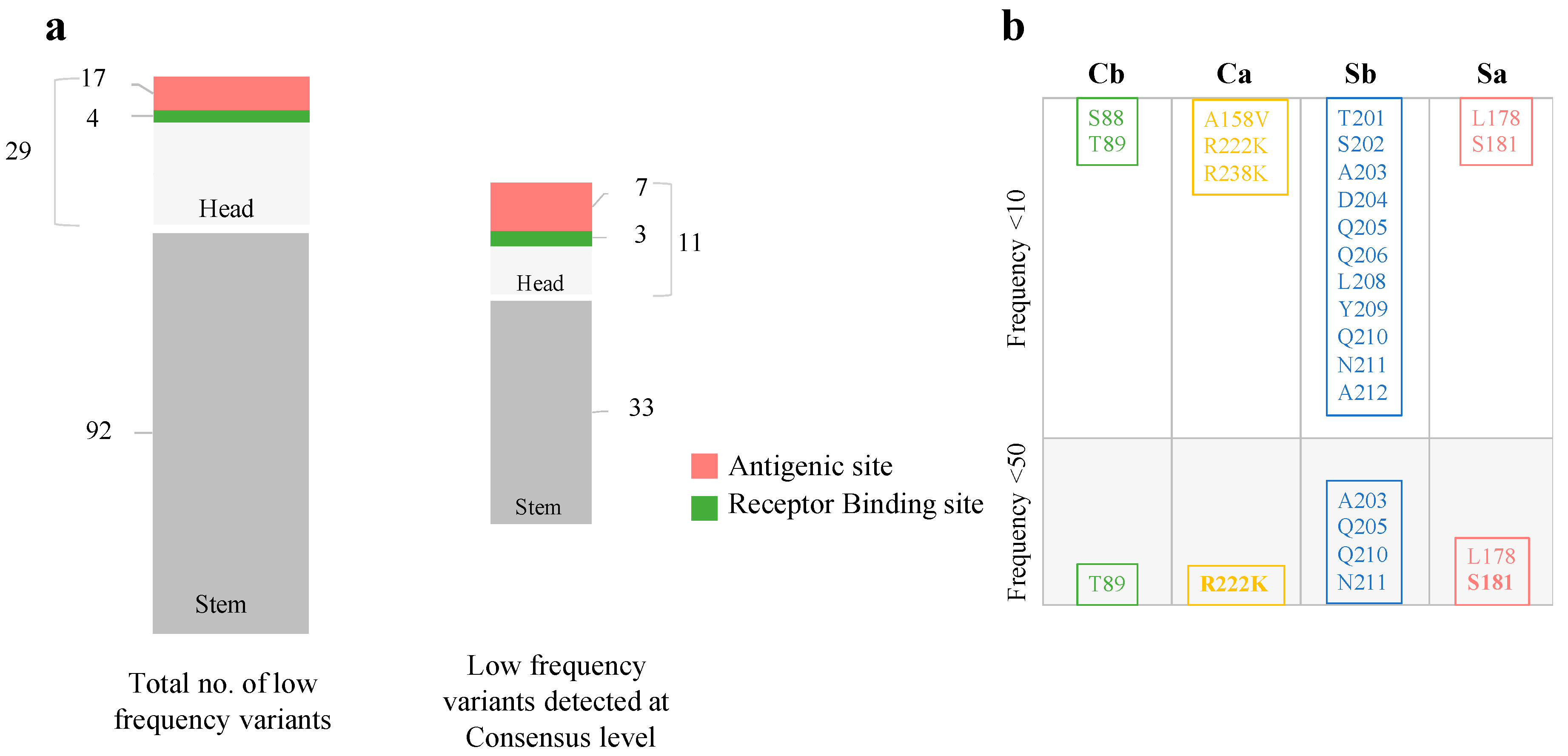
3.3. Beyond the Consensus Sequence: Genetic Diversity of pH1N1 at the Sub-Consensus Level
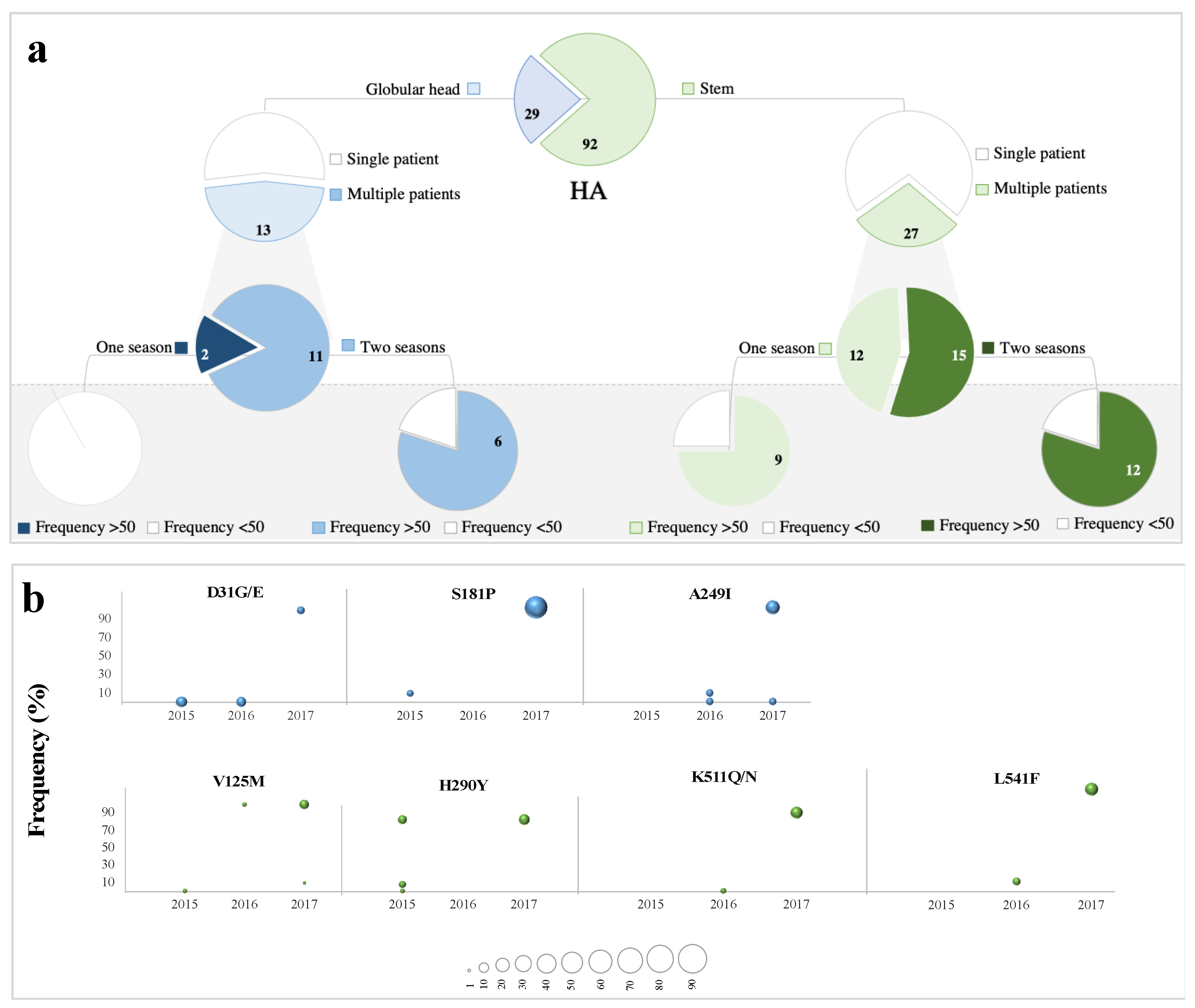
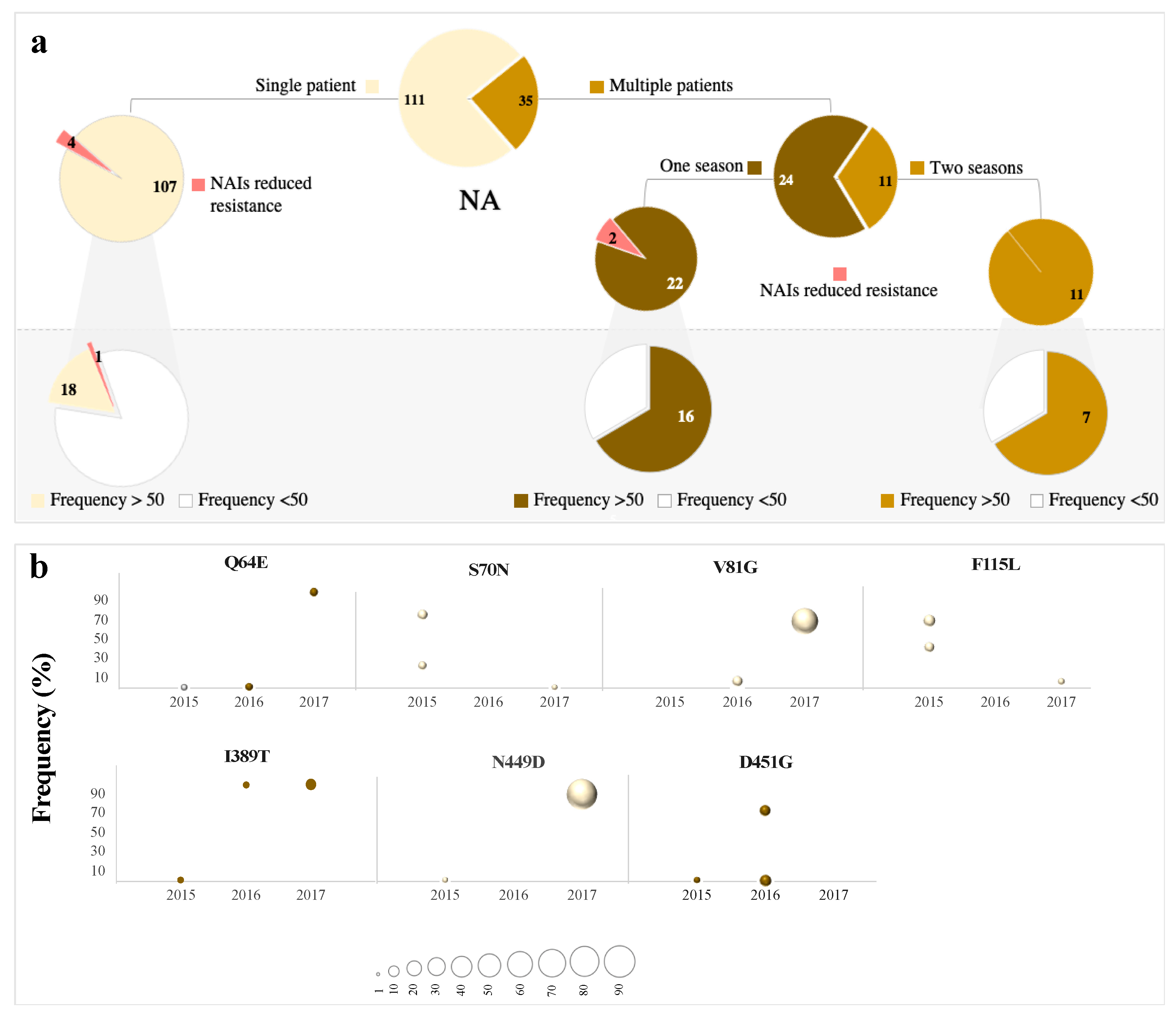
3.4. Low-Frequency Variants in Antigenic Sites of pH1N1 Glycoproteins
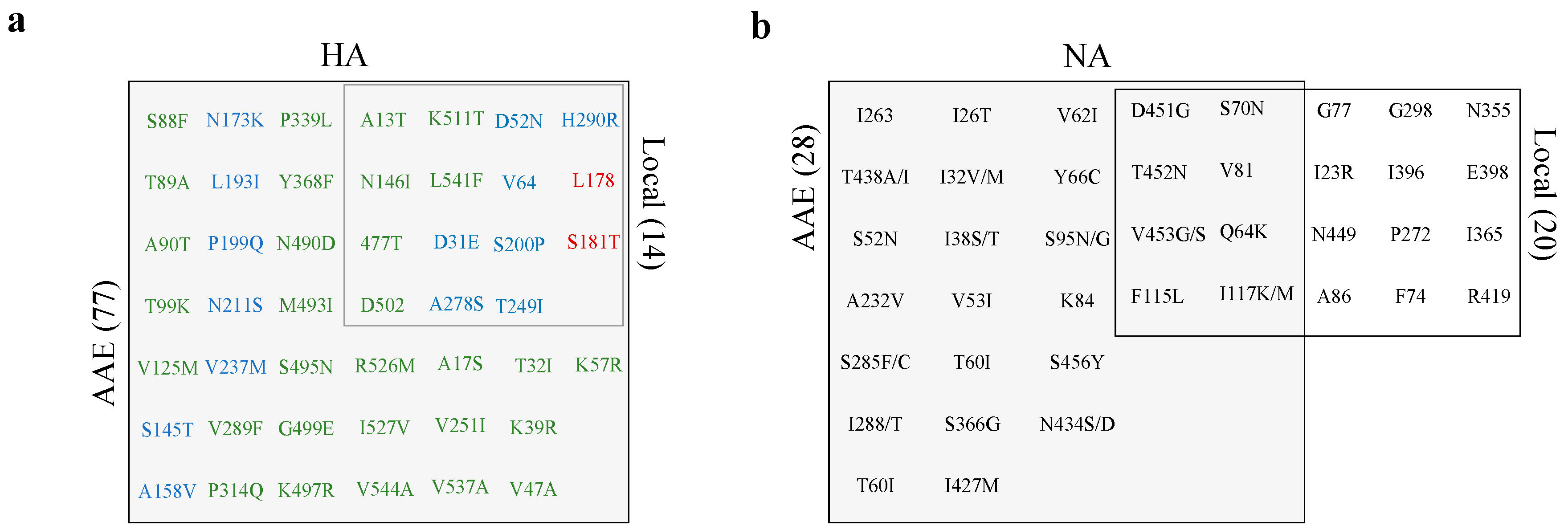
3.5. Abundance is not Necessarily Associated with Emergence at the Consensus Level
3.6. Low-Frequency Variants of HA not NA Repeatedly Arise at the Consensus Level Globally
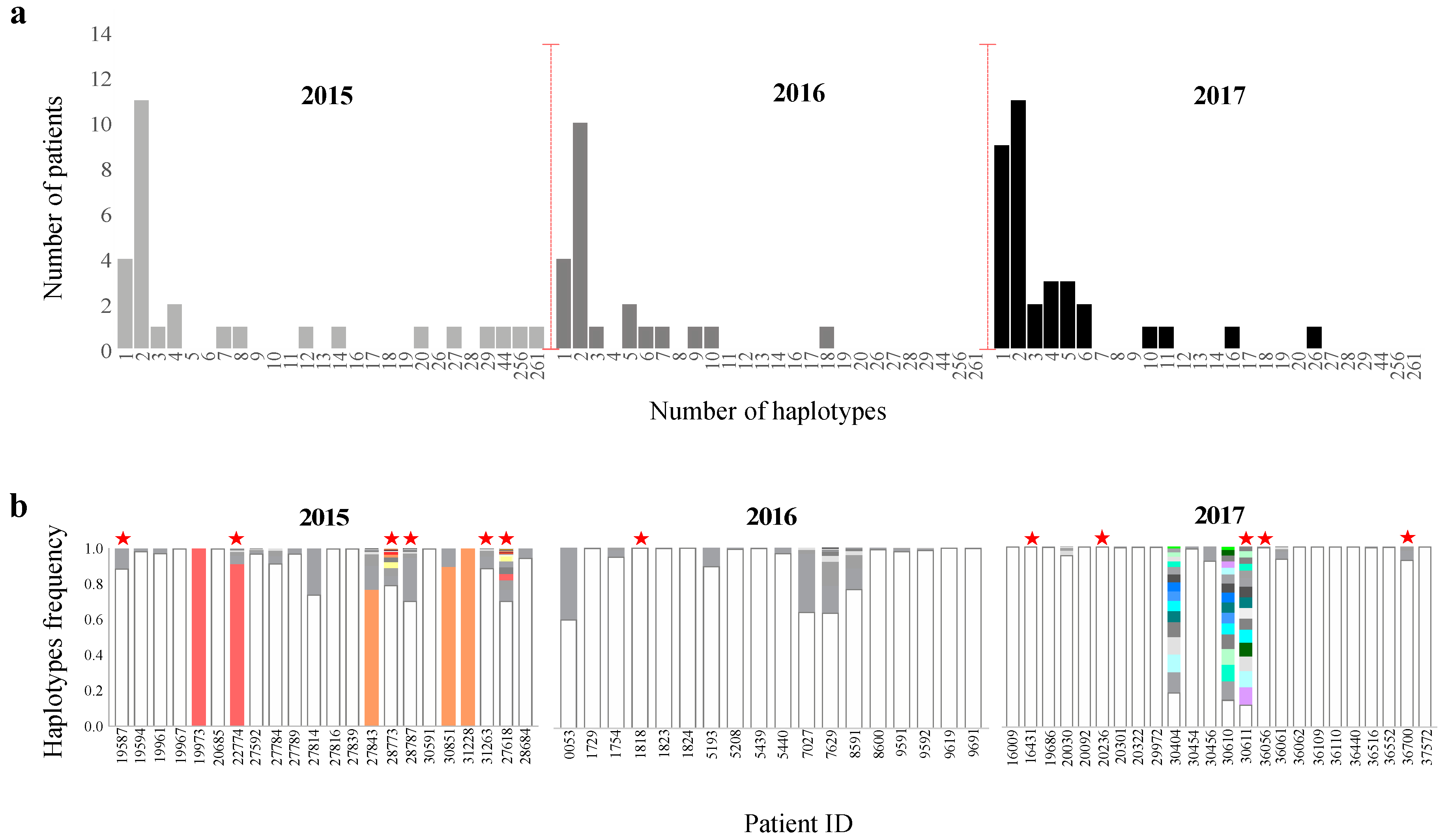
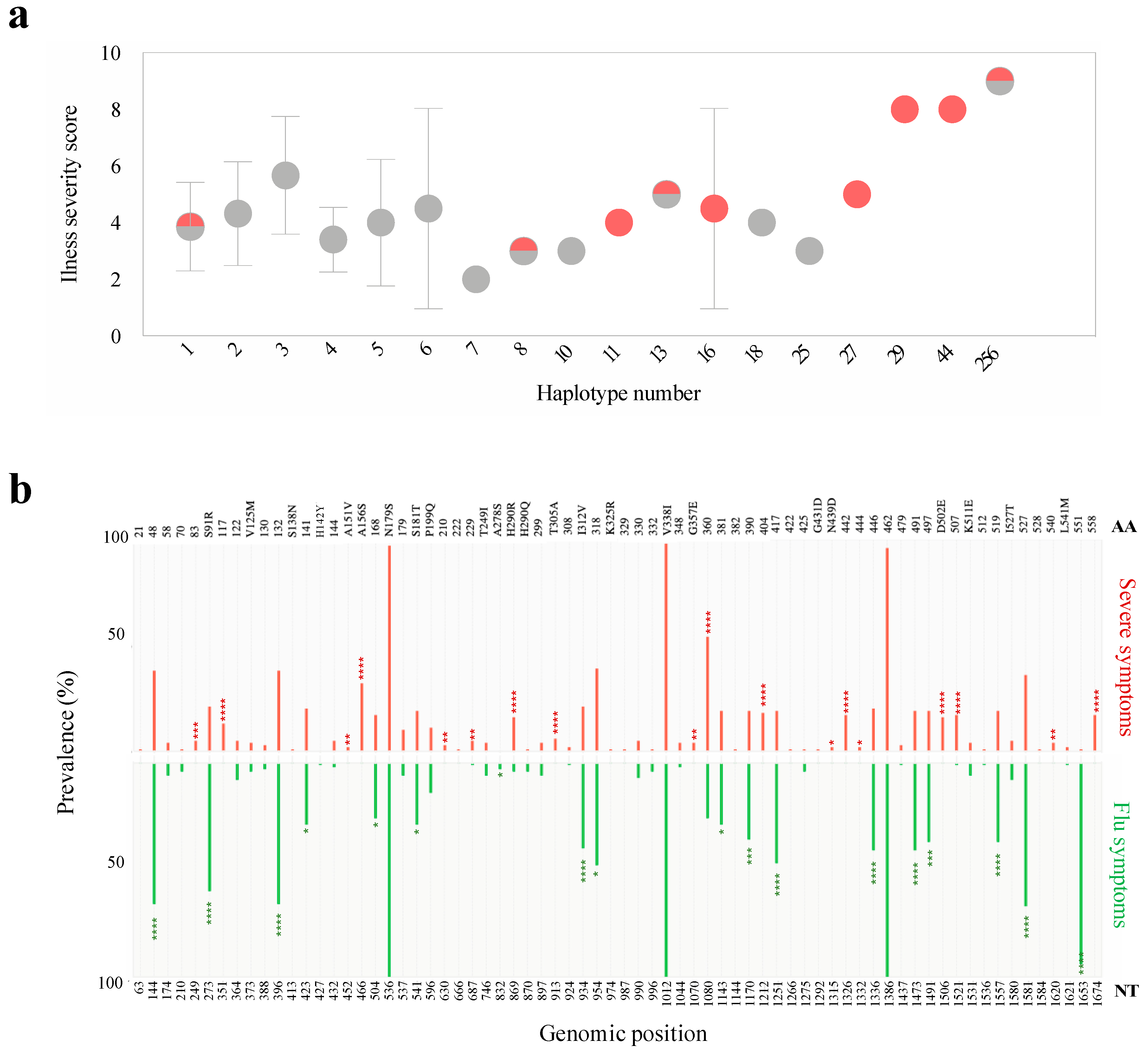
3.7. Scaling up from Single Site Variation to Haplotype Diversity
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Dawood, F.S.; Jain, S.; Finelli, L.; Shaw, M.W.; Lindstrom, S.; Garten, R.J.; Gubareva, L.V.; Xu, X.; Bridges, C.B.; Uyeki, T.M. Emergence of a novel swine-origin influenza A (H1N1) virus in humans. N. Engl. J. Med. 2009, 360, 2605–2615. [Google Scholar] [PubMed] [Green Version]
- WHO. Pandemic (H1N1) 2009-Update 85; World Health Organization: Geneva, Switzerland, 2010. [Google Scholar]
- WHO. H1N1 in Post-Pandemic Period; World Health Organization: Geneva, Switzerland, 2010. [Google Scholar]
- Ghedin, E.; Sengamalay, N.A.; Shumway, M.; Zaborsky, J.; Feldblyum, T.; Subbu, V.; Spiro, D.J.; Sitz, J.; Koo, H.; Bolotov, P.; et al. Large-scale sequencing of human influenza reveals the dynamic nature of viral genome evolution. Nature 2005, 437, 1162–1166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neumann, G.; Noda, T.; Kawaoka, Y. Emergence and pandemic potential of swine-origin H1N1 influenza virus. Nature 2009, 459, 931–939. [Google Scholar] [CrossRef] [Green Version]
- Dias, A.; Bouvier, D.; Crepin, T.; McCarthy, A.A.; Hart, D.J.; Baudin, F.; Cusack, S.; Ruigrok, R.W. The cap-snatching endonuclease of influenza virus polymerase resides in the PA subunit. Nature 2009, 458, 914–918. [Google Scholar] [CrossRef] [PubMed]
- Nobusawa, E.; Sato, K. Comparison of the mutation rates of human influenza A and B viruses. J. Virol. 2006, 80, 3675–3678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lauring, A.S.; Andino, R. Quasispecies theory and the behavior of RNA viruses. PLoS Pathog. 2010, 6, e1001005. [Google Scholar] [CrossRef]
- Holmes, E.C.; Moya, A. Is the quasispecies concept relevant to RNA viruses? J. Virol. 2002, 76, 460–465. [Google Scholar] [CrossRef] [Green Version]
- Domingo, E.; Martinez-Salas, E.; Sobrino, F.; de la Torre, J.C.; Portela, A.; Ortin, J.; Lopez-Galindez, C.; Perez-Brena, P.; Villanueva, N.; Najera, R.; et al. The quasispecies (extremely heterogeneous) nature of viral RNA genome populations: Biological relevance- A review. Gene 1985, 40, 1–8. [Google Scholar] [CrossRef]
- Lemey, P.; Rambaut, A.; Pybus, O.G. HIV evolutionary dynamics within and among hosts. Aids Rev. 2006, 8, 125–140. [Google Scholar]
- Simmonds, P. Genetic diversity and evolution of hepatitis C virus, 15 years on. J. Gen. Virol. 2004, 85, 3173–3188. [Google Scholar] [CrossRef]
- Carrat, F.; Vergu, E.; Ferguson, N.M.; Lemaitre, M.; Cauchemez, S.; Leach, S.; Valleron, A.J. Time lines of infection and disease in human influenza: A review of volunteer challenge studies. Am. J. Epidemiol. 2008, 167, 775–785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCrone, J.T.; Woods, R.J.; Martin, E.T.; Malosh, R.E.; Monto, A.S.; Lauring, A.S. Stochastic processes constrain the within and between host evolution of influenza virus. eLife 2018, 7, e35962. [Google Scholar] [CrossRef] [PubMed]
- Debbink, K.; McCrone, J.T.; Petrie, J.G.; Truscon, R.; Johnson, E.; Mantlo, E.K.; Monto, A.S.; Lauring, A.S. Vaccination has minimal impact on the intrahost diversity of H3N2 influenza viruses. PLoS Pathog. 2017, 13, e1006194. [Google Scholar] [CrossRef] [Green Version]
- Poon, L.L.; Song, T.; Rosenfeld, R.; Lin, X.; Rogers, M.B.; Zhou, B.; Sebra, R.; Halpin, R.A.; Guan, Y.; Twaddle, A.; et al. Quantifying influenza virus diversity and transmission in humans. Nat. Genet. 2016, 48, 195–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luksza, M.; Lassig, M. A predictive fitness model for influenza. Nature 2014, 507, 57–61. [Google Scholar] [CrossRef] [PubMed]
- Varble, A.; Albrecht, R.A.; Backes, S.; Crumiller, M.; Bouvier, N.M.; Sachs, D.; Garcia-Sastre, A. Influenza A virus transmission bottlenecks are defined by infection route and recipient host. Cell Host Microbe 2014, 16, 691–700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grenfell, B.T.; Pybus, O.G.; Gog, J.R.; Wood, J.L.; Daly, J.M.; Mumford, J.A.; Holmes, E.C. Unifying the epidemiological and evolutionary dynamics of pathogens. Science 2004, 303, 327–332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andino, R.; Domingo, E. Viral quasispecies. Virology 2015, 479–480, 46–51. [Google Scholar] [CrossRef] [Green Version]
- Vignuzzi, M.; Stone, J.K.; Arnold, J.J.; Cameron, C.E.; Andino, R. Quasispecies diversity determines pathogenesis through cooperative interactions in a viral population. Nature 2006, 439, 344–348. [Google Scholar] [CrossRef]
- Cheung, P.P.; Watson, S.J.; Choy, K.T.; Fun Sia, S.; Wong, D.D.; Poon, L.L.; Kellam, P.; Guan, Y.; Malik Peiris, J.S.; Yen, H.L. Generation and characterization of influenza A viruses with altered polymerase fidelity. Nat. Commun. 2014, 5, 4794. [Google Scholar] [CrossRef]
- Nowak, M.A.; Anderson, R.M.; McLean, A.R.; Wolfs, T.F.; Goudsmit, J.; May, R.M. Antigenic diversity thresholds and the development of AIDS. Science 1991, 254, 963–969. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.A.; Li, J.F.; Wei, X.; Lipscomb, J.; Irlbeck, D.; Craig, C.; Smith, A.; Bennett, D.E.; Monsour, M.; Sandstrom, P.; et al. Minority HIV-1 drug resistance mutations are present in antiretroviral treatment-naive populations and associate with reduced treatment efficacy. PLoS Med. 2008, 5, e158. [Google Scholar] [CrossRef] [PubMed]
- Dinis, J.M.; Florek, N.W.; Fatola, O.O.; Moncla, L.H.; Mutschler, J.P.; Charlier, O.K.; Meece, J.K.; Belongia, E.A.; Friedrich, T.C. Deep Sequencing Reveals Potential Antigenic Variants at Low Frequencies in Influenza A Virus-Infected Humans. J. Virol. 2016, 90, 3355–3365. [Google Scholar] [CrossRef] [Green Version]
- Duchamp, M.B.; Casalegno, J.S.; Gillet, Y.; Frobert, E.; Bernard, E.; Escuret, V.; Billaud, G.; Valette, M.; Javouhey, E.; Lina, B.; et al. Pandemic A(H1N1)2009 influenza virus detection by real time RT-PCR: Is viral quantification useful? Clin. Microbiol. Infect. 2010, 16, 317–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakauchi, M.; Yasui, Y.; Miyoshi, T.; Minagawa, H.; Tanaka, T.; Tashiro, M.; Kageyama, T. One-step real-time reverse transcription-PCR assays for detecting and subtyping pandemic influenza A/H1N1 2009, seasonal influenza A/H1N1, and seasonal influenza A/H3N2 viruses. J. Virol. Methods 2011, 171, 156–162. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Donnelly, M.E.; Scholes, D.T.; St George, K.; Hatta, M.; Kawaoka, Y.; Wentworth, D.E. Single-reaction genomic amplification accelerates sequencing and vaccine production for classical and Swine origin human influenza a viruses. J. Virol. 2009, 83, 10309–10313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. 2010. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc (accessed on 1 November 2019).
- Krueger, F. A Wrapper Tool around Cutadapt and FastQC to Consistently Apply Quality and Adapter Trimming to FastQ Files. 2015. Available online: https://github.com/FelixKrueger/TrimGalore (accessed on 1 November 2019).
- Gordon, A.H. FastX Toolkit. 2010. Available online: http://hannonlab.cshl.edu/fastx_toolkit/ (accessed on 1 November 2019).
- Al Khatib, H.A.; Al Thani, A.A.; Yassine, H.M. Evolution and dynamics of the pandemic H1N1 influenza hemagglutinin protein from 2009 to 2017. Arch. Virol. 2018, 163, 3035–3049. [Google Scholar] [CrossRef]
- Van den Hoecke, S.; Verhelst, J.; Vuylsteke, M.; Saelens, X. Analysis of the genetic diversity of influenza A viruses using next-generation DNA sequencing. BMC Genom. 2015, 16, 79. [Google Scholar] [CrossRef] [Green Version]
- Sobel Leonard, A.; McClain, M.T.; Smith, G.J.; Wentworth, D.E.; Halpin, R.A.; Lin, X.; Ransier, A.; Stockwell, T.B.; Das, S.R.; Gilbert, A.S.; et al. Deep Sequencing of Influenza A Virus from a Human Challenge Study Reveals a Selective Bottleneck and Only Limited Intrahost Genetic Diversification. J. Virol. 2016, 90, 11247–11258. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilm, A.; Aw, P.P.; Bertrand, D.; Yeo, G.H.; Ong, S.H.; Wong, C.H.; Khor, C.C.; Petric, R.; Hibberd, M.L.; Nagarajan, N. LoFreq: A sequence-quality aware, ultra-sensitive variant caller for uncovering cell-population heterogeneity from high-throughput sequencing datasets. Nucleic Acids Res. 2012, 40, 11189–11201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef] [PubMed]
- Hall, T.A. BioEdit: A user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 1999, 41, 95–98. [Google Scholar]
- Drummond, A.J.; Rambaut, A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 2007, 7, 214. [Google Scholar] [CrossRef] [Green Version]
- Drummond, A.J.; Suchard, M.A.; Xie, D.; Rambaut, A. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol. Biol. Evol. 2012, 29, 1969–1973. [Google Scholar] [CrossRef] [Green Version]
- Bednarska, K.; Hallmann-Szelinska, E.; Kondratiuk, K.; Brydak, L.B. Evaluation of the Activity of Influenza and Influenza-Like Viruses in the Epidemic Season 2013/2014. Adv. Exp. Med. Biol. 2015, 857, 1–7. [Google Scholar] [CrossRef]
- Pond, S.L.; Frost, S.D.; Muse, S.V. HyPhy: Hypothesis testing using phylogenies. Bioinformatics 2005, 21, 676–679. [Google Scholar] [CrossRef] [Green Version]
- Routh, A.; Chang, M.W.; Okulicz, J.F.; Johnson, J.E.; Torbett, B.E. CoVaMa: Co-Variation Mapper for disequilibrium analysis of mutant loci in viral populations using next-generation sequence data. Methods 2015, 91, 40–47. [Google Scholar] [CrossRef] [Green Version]
- Topfer, A.; Zagordi, O.; Prabhakaran, S.; Roth, V.; Halperin, E.; Beerenwinkel, N. Probabilistic inference of viral quasispecies subject to recombination. J. Comput. Biol. 2013, 20, 113–123. [Google Scholar] [CrossRef]
- Capelastegui, A.; Quintana, J.M.; Bilbao, A.; Espana, P.P.; Garin, O.; Alonso, J.; Astray, J.; Canton, R.; Castilla, J.; Castro, A.; et al. Score to identify the severity of adult patients with influenza A (H1N1) 2009 virus infection at hospital admission. Eur. J. Clin. Microbiol. Infect. Dis. 2012, 31, 2693–2701. [Google Scholar] [CrossRef] [PubMed]
- Lo, C.Y.; Tang, Y.S.; Shaw, P.C. Structure and Function of Influenza Virus Ribonucleoprotein. Subcell. Biochem. 2018, 88, 95–128. [Google Scholar] [PubMed]
- WHO. Weekly Seasonal Influenza Updates; World Health Organization: Geneva, Switzerland, 2016. [Google Scholar]
- WHO. Summary of Neuraminidase Amino Acid Substitutions Associated with Reduced Inhibition by Neuraminidase Inhibitors (NAI); World Health Organization: Geneva, Switzerland, 2016. [Google Scholar]
- Bhoye, D.; Behera, A.K.; Cherian, S.S. A molecular modelling approach to understand the effect of co-evolutionary mutations (V344M, I354L) identified in the PB2 subunit of influenza A 2009 pandemic H1N1 virus on m7GTP ligand binding. J. Gen. Virol. 2016, 97, 1785–1796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laplante, J.; St George, K. Antiviral resistance in influenza viruses: Laboratory testing. Clin. Lab. Med. 2014, 34, 387–408. [Google Scholar] [CrossRef]
- World Health Organisation. Influenza Collaberating Center-London-Annual and Interim Reports; World Health Organization: Geneva, Switzerland, 2009–2017. [Google Scholar]
- Xue, K.S.; Moncla, L.H.; Bedford, T.; Bloom, J.D. Within-Host Evolution of Human Influenza Virus. Trends Microbiol. 2018, 26, 781–793. [Google Scholar] [CrossRef]
- Koel, B.F.; Burke, D.F.; Bestebroer, T.M.; van der Vliet, S.; Zondag, G.C.; Vervaet, G.; Skepner, E.; Lewis, N.S.; Spronken, M.I.; Russell, C.A.; et al. Substitutions near the receptor binding site determine major antigenic change during influenza virus evolution. Science 2013, 342, 976–979. [Google Scholar] [CrossRef]
- Van der Vries, E.; Schutten, M.; Fraaij, P.; Boucher, C.; Osterhaus, A. Influenza virus resistance to antiviral therapy. Adv. Pharmacol. 2013, 67, 217–246. [Google Scholar]
- Renaud, C.; Kuypers, J.; Englund, J.A. Emerging oseltamivir resistance in seasonal and pandemic influenza A/H1N1. J. Clin. Virol. 2011, 52, 70–78. [Google Scholar] [CrossRef]
- Ambati, A.; Valentini, D.; Montomoli, E.; Lapini, G.; Biuso, F.; Wenschuh, H.; Magalhaes, I.; Maeurer, M. H1N1 viral proteome peptide microarray predicts individuals at risk for H1N1 infection and segregates infection versus Pandemrix((R)) vaccination. Immunology 2015, 145, 357–366. [Google Scholar] [CrossRef]
- Gopal, R.; Rangel-Moreno, J.; Fallert Junecko, B.A.; Mallon, D.J.; Chen, K.; Pociask, D.A.; Connell, T.D.; Reinhart, T.A.; Alcorn, J.F.; Ross, T.M.; et al. Mucosal pre-exposure to Th17-inducing adjuvants exacerbates pathology after influenza infection. Am. J. Pathol. 2014, 184, 55–63. [Google Scholar] [CrossRef] [Green Version]
- Clark, A.M.; Nogales, A.; Martinez-Sobrido, L.; Topham, D.J.; DeDiego, M.L. Functional Evolution of Influenza Virus NS1 Protein in Currently Circulating Human 2009 Pandemic H1N1 Viruses. J. Virol. 2017, 91, e00721-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krug, R.M. Functions of the influenza A virus NS1 protein in antiviral defense. Curr. Opin. Virol. 2015, 12, 1–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Itoh, Y.; Shinya, K.; Kiso, M.; Watanabe, T.; Sakoda, Y.; Hatta, M.; Muramoto, Y.; Tamura, D.; Sakai-Tagawa, Y.; Noda, T.; et al. In vitro and in vivo characterization of new swine-origin H1N1 influenza viruses. Nature 2009, 460, 1021–1025. [Google Scholar] [CrossRef] [PubMed]
- Rehwinkel, J.; Tan, C.P.; Goubau, D.; Schulz, O.; Pichlmair, A.; Bier, K.; Robb, N.; Vreede, F.; Barclay, W.; Fodor, E.; et al. RIG-I detects viral genomic RNA during negative-strand RNA virus infection. Cell 2010, 140, 397–408. [Google Scholar] [CrossRef] [Green Version]
- Hale, B.G.; Jackson, D.; Chen, Y.H.; Lamb, R.A.; Randall, R.E. Influenza A virus NS1 protein binds p85beta and activates phosphatidylinositol-3-kinase signaling. Proc. Natl. Acad. Sci. USA 2006, 103, 14194–14199. [Google Scholar] [CrossRef] [Green Version]
- Viboud, C.; Nelson, M.I.; Tan, Y.; Holmes, E.C. Contrasting the epidemiological and evolutionary dynamics of influenza spatial transmission. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2013, 368, 20120199. [Google Scholar] [CrossRef] [Green Version]
- Nowak, M.A. What is a quasispecies? Trends Ecol. Evol. 1992, 7, 118–121. [Google Scholar] [CrossRef]
- Drake, J.W.; Holland, J.J. Mutation rates among RNA viruses. Proc. Natl. Acad. Sci. USA 1999, 96, 13910–13913. [Google Scholar] [CrossRef] [Green Version]
- Zagordi, O.; Daumer, M.; Beisel, C.; Beerenwinkel, N. Read length versus depth of coverage for viral quasispecies reconstruction. PLoS ONE 2012, 7, e47046. [Google Scholar] [CrossRef] [Green Version]
- Xue, K.S.; Stevens-Ayers, T.; Campbell, A.P.; Englund, J.A.; Pergam, S.A.; Boeckh, M.; Bloom, J.D. Parallel evolution of influenza across multiple spatiotemporal scales. eLife 2017, 6, e26875. [Google Scholar] [CrossRef]
- Su, Y.C.; Bahl, J.; Joseph, U.; Butt, K.M.; Peck, H.A.; Koay, E.S.; Oon, L.L.; Barr, I.G.; Vijaykrishna, D.; Smith, G.J. Phylodynamics of H1N1/2009 influenza reveals the transition from host adaptation to immune-driven selection. Nat. Commun. 2015, 6, 7952. [Google Scholar] [CrossRef] [Green Version]
- Guarnaccia, T.; Carolan, L.A.; Maurer-Stroh, S.; Lee, R.T.; Job, E.; Reading, P.C.; Petrie, S.; McCaw, J.M.; McVernon, J.; Hurt, A.C.; et al. Antigenic drift of the pandemic 2009 A(H1N1) influenza virus in A ferret model. PLoS Pathog. 2013, 9, e1003354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwatsuki-Horimoto, K.; Horimoto, T.; Fujii, Y.; Kawaoka, Y. Generation of influenza A virus NS2 (NEP) mutants with an altered nuclear export signal sequence. J. Virol. 2004, 78, 10149–10155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moncla, L.H.; Zhong, G.; Nelson, C.W.; Dinis, J.M.; Mutschler, J.; Hughes, A.L.; Watanabe, T.; Kawaoka, Y.; Friedrich, T.C. Selective bottlenecks shape evolutionary pathways taken during mammalian adaptation of a 1918-like avian influenza virus. Cell Host Microbe 2016, 19, 169–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Georgieva, M.; Buckee, C.O.; Lipsitch, M. Models of immune selection for multi-locus antigenic diversity of pathogens. Nat. Rev. 2019, 19, 55–62. [Google Scholar] [CrossRef]
- Hensley, S.E.; Das, S.R.; Bailey, A.L.; Schmidt, L.M.; Hickman, H.D.; Jayaraman, A.; Viswanathan, K.; Raman, R.; Sasisekharan, R.; Bennink, J.R.; et al. Hemagglutinin receptor binding avidity drives influenza A virus antigenic drift. Science 2009, 326, 734–736. [Google Scholar] [CrossRef] [Green Version]









© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Al Khatib, H.A.; Al Maslamani, M.A.; Coyle, P.V.; Thompson, I.R.; Farag, E.A.; Al Thani, A.A.; Yassine, H.M. Inter-Versus Intra-Host Sequence Diversity of pH1N1 and Associated Clinical Outcomes. Microorganisms 2020, 8, 133. https://doi.org/10.3390/microorganisms8010133
Al Khatib HA, Al Maslamani MA, Coyle PV, Thompson IR, Farag EA, Al Thani AA, Yassine HM. Inter-Versus Intra-Host Sequence Diversity of pH1N1 and Associated Clinical Outcomes. Microorganisms. 2020; 8(1):133. https://doi.org/10.3390/microorganisms8010133
Chicago/Turabian StyleAl Khatib, Hebah A., Muna A. Al Maslamani, Peter V. Coyle, I. Richard Thompson, Elmoubasher A. Farag, Asmaa A. Al Thani, and Hadi M. Yassine. 2020. "Inter-Versus Intra-Host Sequence Diversity of pH1N1 and Associated Clinical Outcomes" Microorganisms 8, no. 1: 133. https://doi.org/10.3390/microorganisms8010133