Chronic Inflammatory Diseases at Secondary Sites Ensuing Urogenital or Pulmonary Chlamydia Infections


Abstract
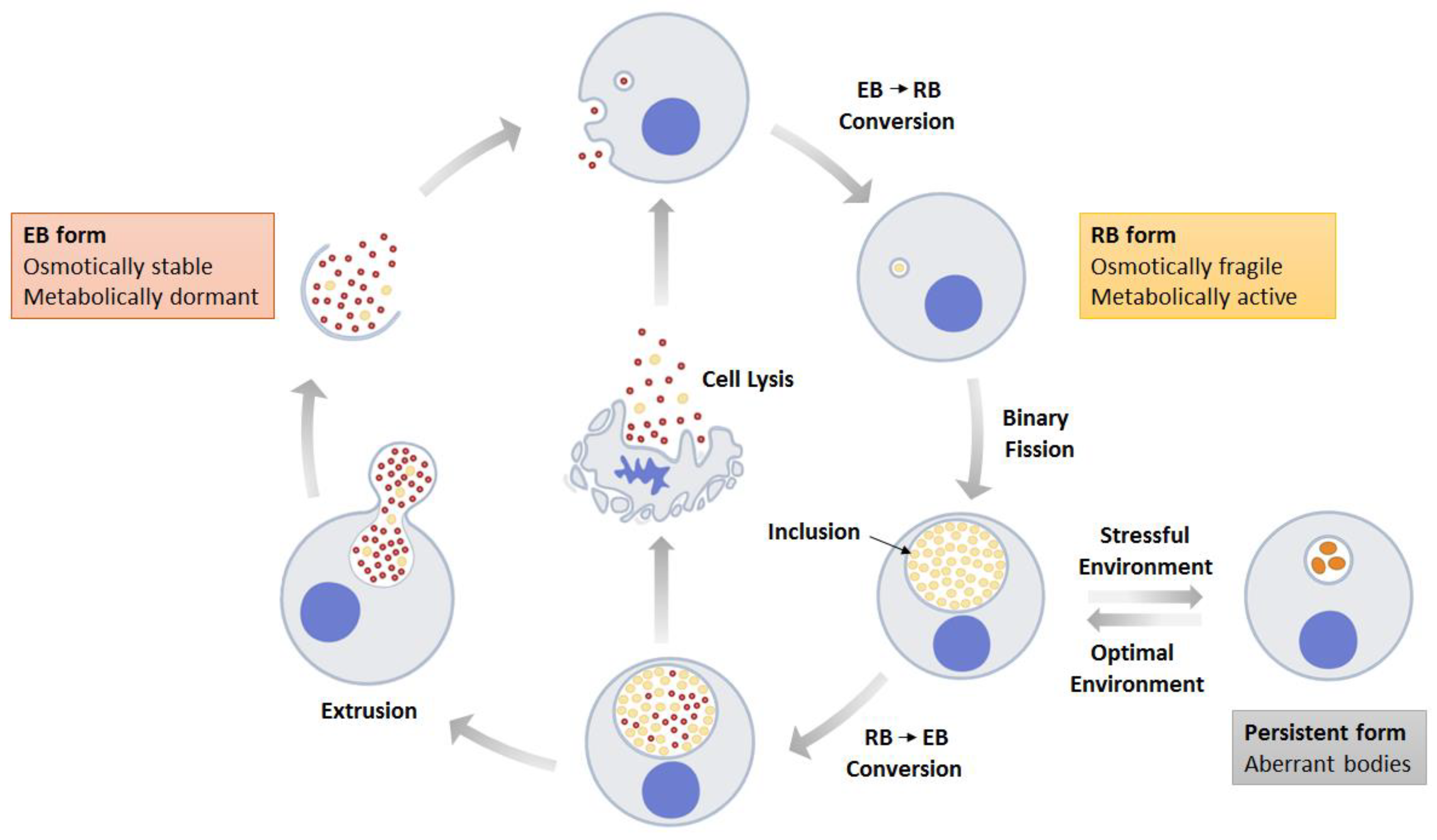
:1. Introduction
2. Chlamydial Infection at the Primary Sites
3. C. trachomatis and Reactive Arthritis
3.1. Bacterial Travel Beyond Urogenital Tract, Hypoxic Environment, and Persistence Stage Fuel the Occurrence of Post-Venereal Reactive Arthritis
3.2. Molecular Mimicry and Inflammatory Property of Chlamydial Antigens Triggers Local Inflammatory Response in Reactive Arthritis
3.3. Antibiotic Treatment for Reactive Arthritis
4. C. pneumoniae and Atherosclerosis
4.1. Adhesive C. pneumoniae-Infected Monocytes and Foam Cell to Endothelium Enhances Atherosclerotic Plaque Formation
4.2. TLR4- and MyD88-Dependent Host Immune Response Accelerates Chlamydial-Mediated Atherosclerosis
4.3. Platelet Aggregation by C. pneumoniae Enhances Atherosclerotic Plaque Formation
5. C. pneumoniae and Multiple Sclerosis
5.1. C. pneumoniae Infects Brain Endothelial Cells and Disrupts Integrity of Blood-Brain Barrier
5.2. Molecular Mimicry
6. C. pneumoniae and Alzheimer’s Disease
6.1. C. pneumoniae Infected Monocytes Gain Penetration through Brain Endothelial Cells
6.2. Inflammatory LPS-Induced Activation of Microglial
6.3. Antibiotic Treatment for Alzheimer’s Disease
7. C. pneumoniae and Asthma
8. C. pneumoniae and Primary Biliary Cirrhosis
9. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Brunham, R.C.; Rey-Ladino, J. Immunology of Chlamydia infection: Implications for a Chlamydia trachomatis vaccine. Nat. Rev. Immunol 2005, 5, 149–161. [Google Scholar] [CrossRef] [PubMed]
- Bavoil, P.; Ohlin, A.; Schachter, J. Role of disulfide bonding in outer membrane structure and permeability in Chlamydia trachomatis. Infect. Immun. 1984, 44, 479–485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Betts-Hampikian, H.J.; Fields, K.A. Disulfide bonding within components of the Chlamydia type III secretion apparatus correlates with development. J. Bacteriol. 2011, 193, 6950–6959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.K.; Enciso, G.A.; Boassa, D.; Chander, C.N.; Lou, T.H.; Pairawan, S.S.; Guo, M.C.; Wan, F.Y.M.; Ellisman, M.H.; Sutterlin, C.; et al. Replication-dependent size reduction precedes differentiation in Chlamydia trachomatis. Nat. Commun. 2018, 9, 45. [Google Scholar] [CrossRef]
- Hybiske, K.; Stephens, R.S. Mechanisms of host cell exit by the intracellular bacterium Chlamydia. Proc. Natl. Acad. Sci. USA 2007, 104, 11430–11435. [Google Scholar] [CrossRef] [Green Version]
- Panzetta, M.E.; Valdivia, R.H.; Saka, H.A. Chlamydia Persistence: A Survival Strategy to Evade Antimicrobial Effects in-vitro and in-vivo. Front. Microbiol. 2018, 9, 3101. [Google Scholar] [CrossRef]
- Belland, R.J.; Nelson, D.E.; Virok, D.; Crane, D.D.; Hogan, D.; Sturdevant, D.; Beatty, W.L.; Caldwell, H.D. Transcriptome analysis of chlamydial growth during IFN-gamma-mediated persistence and reactivation. Proc. Natl. Acad. Sci. USA 2003, 100, 15971–15976. [Google Scholar] [CrossRef] [Green Version]
- Gerard, H.C.; Krausse-Opatz, B.; Wang, Z.; Rudy, D.; Rao, J.P.; Zeidler, H.; Schumacher, H.R.; Whittum-Hudson, J.A.; Kohler, L.; Hudson, A.P. Expression of Chlamydia trachomatis genes encoding products required for DNA synthesis and cell division during active versus persistent infection. Mol. Microbiol. 2001, 41, 731–741. [Google Scholar] [CrossRef]
- Brinkworth, A.J.; Wildung, M.R.; Carabeo, R.A. Genomewide Transcriptional Responses of Iron-Starved Chlamydia trachomatis Reveal Prioritization of Metabolic Precursor Synthesis over Protein Translation. mSystems 2018, 3. [Google Scholar] [CrossRef] [Green Version]
- Tan, G.M.; Lim, H.J.; Yeow, T.C.; Movahed, E.; Looi, C.Y.; Gupta, R.; Arulanandam, B.P.; Abu Bakar, S.; Sabet, N.S.; Chang, L.Y.; et al. Temporal proteomic profiling of Chlamydia trachomatis-infected HeLa-229 human cervical epithelial cells. Proteomics 2016, 16, 1347–1360. [Google Scholar] [CrossRef]
- Olive, A.J.; Haff, M.G.; Emanuele, M.J.; Sack, L.M.; Barker, J.R.; Elledge, S.J.; Starnbach, M.N. Chlamydia trachomatis-induced alterations in the host cell proteome are required for intracellular growth. Cell Host Microbe 2014, 15, 113–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, W.F.; Chambers, J.P.; Gupta, R.; Arulanandam, B.P. Chlamydia and Its Many Ways of Escaping the Host Immune System. J. Pathog. 2019, 2019, 8604958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keat, A.C.; Thomas, B.J.; Taylor-Robinson, D.; Pegrum, G.D.; Maini, R.N.; Scott, J.T. Evidence of Chlamydia trachomatis infection in sexually acquired reactive arthritis. Ann. Rheum. Dis. 1980, 39, 431–437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vilppula, A.H.; Yli-Kerttula, U.I.; Ahlroos, A.K.; Terho, P.E. Chlamydial isolations and serology in Reiter’s syndrome. Scand. J. Rheumatol. 1981, 10, 181–185. [Google Scholar] [CrossRef] [PubMed]
- Zeidler, H.; Kuipers, J.; Köhler, L. Chlamydia-induced arthritis. Curr. Opin. Rheumatol. 2004, 16, 380–392. [Google Scholar] [CrossRef]
- Moazed, T.C.; Kuo, C.-C.; Grayston, J.T.; Campbell, L.A. Evidence of Systemic Dissemination of Chlamydia pneumoniae via Macrophages in the Mouse. J. Infect. Dis. 1998, 177, 1322–1325. [Google Scholar] [CrossRef] [Green Version]
- Gieffers, J.; van Zandbergen, G.; Rupp, J.; Sayk, F.; Krüger, S.; Ehlers, S.; Solbach, W.; Maass, M. Phagocytes transmit Chlamydia pneumoniae from the lungs to the vasculature. Eur. Respir. J. 2004, 23, 506–510. [Google Scholar] [CrossRef] [Green Version]
- Roth, A.; König, P.; van Zandbergen, G.; Klinger, M.; Hellwig-Bürgel, T.; Däubener, W.; Bohlmann, M.K.; Rupp, J. Hypoxia abrogates antichlamydial properties of IFN-γ in human fallopian tube cells in vitro and ex vivo. Proc. Natl. Acad. Sci. USA 2010, 107, 19502–19507. [Google Scholar] [CrossRef] [Green Version]
- Gracey, E.; Inman, R.D. Chlamydia-induced ReA: Immune imbalances and persistent pathogens. Nat. Rev. Rheumatol. 2011, 8, 55. [Google Scholar] [CrossRef]
- Beatty, W.L.; Belanger, T.A.; Desai, A.A.; Morrison, R.P.; Byrne, G.I. Tryptophan depletion as a mechanism of gamma interferon-mediated chlamydial persistence. Infect. Immun. 1994, 62, 3705–3711. [Google Scholar] [CrossRef] [Green Version]
- Gaston, J.S.; Deane, K.H.; Jecock, R.M.; Pearce, J.H. Identification of 2 Chlamydia trachomatis antigens recognized by synovial fluid T cells from patients with Chlamydia induced reactive arthritis. J. Rheumatol. 1996, 23, 130–136. [Google Scholar] [PubMed]
- Hassell, A.B.; Reynolds, D.J.; Deacon, M.; Gaston, J.S.; Pearce, J.H. Identification of T-cell stimulatory antigens of Chlamydia trachomatis using synovial fluid-derived T-cell clones. Immunology 1993, 79, 513–519. [Google Scholar] [PubMed]
- Yazouli, L.E.; Hejaji, H.; Elmdaghri, N.; Alami, A.A.; Dakka, N.; Radouani, F. Investigation of Chlamydia pneumoniae infection in Moroccan patients suffering from cardiovascular diseases. J. Infect. Public Health 2018, 11, 246–249. [Google Scholar] [CrossRef] [PubMed]
- Tumurkhuu, G.; Dagvadorj, J.; Porritt, R.A.; Crother, T.R.; Shimada, K.; Tarling, E.J.; Erbay, E.; Arditi, M.; Chen, S. Chlamydia pneumoniae Hijacks a Host Autoregulatory IL-1beta Loop to Drive Foam Cell Formation and Accelerate Atherosclerosis. Cell Metab. 2018, 28, 432–448. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Shimada, K.; Crother, T.R.; Erbay, E.; Shah, P.K.; Arditi, M. Chlamydia and Lipids Engage a Common Signaling Pathway That Promotes Atherogenesis. J. Am. Coll. Cardiol. 2018, 71, 1553–1570. [Google Scholar] [CrossRef]
- Evani, S.J.; Ramasubramanian, A.K. Biophysical regulation of Chlamydia pneumoniae-infected monocyte recruitment to atherosclerotic foci. Sci. Rep. 2016, 6, 19058. [Google Scholar] [CrossRef] [Green Version]
- Zafiratos, M.T.; Manam, S.; Henderson, K.K.; Ramsey, K.H.; Murthy, A.K. CD8+ T cells mediate Chlamydia pneumoniae-induced atherosclerosis in mice. Pathog. Dis. 2015, 73, ftv052. [Google Scholar] [CrossRef]
- Kälvegren, H.; Bylin, H.; Leanderson, P.; Richter, A.; Grenegård, M.; Bengtsson, T. Chlamydia pneumoniae induces nitric oxide synthase and lipoxygenase-dependent production of reactive oxygen species in platelets—Effects on oxidation of low-density lipoproteins. Thromb. Haemost. 2005, 94, 327–335. [Google Scholar] [CrossRef] [Green Version]
- Kälvegren, H.; Majeed, M.; Bengtsson, T. Chlamydia pneumoniae binds to platelets and triggers P-Selectin expression and aggregation. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 1677–1683. [Google Scholar] [CrossRef] [Green Version]
- Sriram, S.; Mitchell, W.; Stratton, C. Multiple sclerosis associated with Chlamydia pneumoniae infection of the CNS. Neurology 1998, 50, 571–572. [Google Scholar] [CrossRef]
- Sriram, S.; Stratton, C.W.; Yao, S.-Y.; Tharp, A.; Ding, L.; Bannan, J.D.; Mitchell, W.M. Chlamydia pneumoniae infection of the central nervous system in multiple sclerosis. Ann. Neurol. 1999, 46, 6–14. [Google Scholar] [CrossRef]
- Layh-Schmitt, G.; Bendl, C.; Hildt, U.; Dong-Si, T.; Jüttler, E.; Schnitzler, P.; Grond-Ginsbach, C.; Grau, A.J. Evidence for infection with Chlamydia pneumoniae in a subgroup of patients with multiple sclerosis. Ann. Neurol. 2000, 47, 652–655. [Google Scholar] [CrossRef]
- Giovannoni, G.; Cutter, G.R.; Lunemann, J.; Martin, R.; Münz, C.; Sriram, S.; Steiner, I.; Hammerschlag, M.R.; Gaydos, C.A. Infectious causes of multiple sclerosis. Lancet Neurol. 2006, 5, 887–894. [Google Scholar] [CrossRef]
- Munger, K.L.; Peeling, R.W.; Hernán, M.A.; Chasan-Taber, L.; Olek, M.J.; Hankinson, S.E.; Hunter, D.; Ascherio, A. Infection with Chlamydia pneumoniae and Risk of Multiple Sclerosis. Epidemiology 2003, 14, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Hammerschlag, M.R.; Ke, Z.; Lu, F.; Roblin, P.; Boman, J.; Kalman, B. Is Chlamydia pneumoniae Present in Brain Lesions of Patients with Multiple Sclerosis? J. Clin. Microbiol. 2000, 38, 4274–4276. [Google Scholar] [CrossRef] [Green Version]
- Gieffers, J.; Pohl, D.; Treib, J.; Dittmann, R.; Stephan, C.; Klotz, K.; Hanefeld, F.; Solbach, W.; Haass, A.; Maass, M. Presence of Chlamydia pneumoniae DNA in the cerebral spinal fluid is a common phenomenon in a variety of neurological diseases and not restricted to multiple sclerosis. Ann. Neurol. 2001, 49, 585–589. [Google Scholar] [CrossRef]
- MacIntyre, A.; Appelt, D.M.; Little, C.S.; Hammond, C.J.; Balin, B.J. Chlamydia pneumoniae infection alters the junctional complex proteins of human brain microvascular endothelial cells. FEMS Microbiol. Lett. 2002, 217, 167–172. [Google Scholar] [CrossRef] [Green Version]
- Molestina, R.E.; Miller, R.D.; Ramirez, J.A.; Summersgill, J.T. Infection of Human Endothelial Cells with Chlamydia pneumoniae Stimulates Transendothelial Migration of Neutrophils and Monocytes. Infect. Immun. 1999, 67, 1323–1330. [Google Scholar] [CrossRef] [Green Version]
- Boelen, E.; Steinbusch, H.W.M.; van der Ven, A.J.A.M.; Grauls, G.; Bruggeman, C.A.; Stassen, F.R.M. Chlamydia pneumoniae infection of brain cells: An in vitro study. Neurobiol. Aging 2007, 28, 524–532. [Google Scholar] [CrossRef]
- Cappello, F.; Conway de Macario, E.; Di Felice, V.; Zummo, G.; Macario, A.J.L. Chlamydia trachomatis infection and anti-Hsp60 immunity: The two sides of the coin. PLoS Pathog. 2009, 5, e1000552. [Google Scholar] [CrossRef] [Green Version]
- Lenz, D.C.; Lu, L.; Conant, S.B.; Wolf, N.A.; Gérard, H.C.; Whittum-Hudson, J.A.; Hudson, A.P.; Swanborg, R.H. A Chlamydia pneumoniae-Specific Peptide Induces Experimental Autoimmune Encephalomyelitis in Rats. J. Immunol. 2001, 167, 1803–1808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balin, B.J.; Gérard, H.C.; Arking, E.J.; Appelt, D.M.; Branigan, P.J.; Abrams, J.T.; Whittum-Hudson, J.A.; Hudson, A.P. Identification and localization of Chlamydia pneumoniae in the Alzheimer’s brain. Med. Microbiol. Immunol. 1998, 187, 23–42. [Google Scholar] [CrossRef] [PubMed]
- Hammond, C.J.; Hallock, L.R.; Howanski, R.J.; Appelt, D.M.; Little, C.S.; Balin, B.J. Immunohistological detection of Chlamydia pneumoniae in the Alzheimer’s disease brain. BMC Neurosci. 2010, 11, 121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oszust, C.; Gérard, H.C.; Whittum-Hudson, J.A.; Wildt, K.S.; Deka, S.; Dreses-Werringloer, U.; Hudson, A.P.; Balin, B.J.; Bordayo, E.Z.; Frey, W.H., II. Chlamydophila (Chlamydia) pneumoniae in the Alzheimer’s brain. FEMS Immunol. Med. Microbiol. 2006, 48, 355–366. [Google Scholar] [CrossRef] [Green Version]
- Little, C.S.; Hammond, C.J.; MacIntyre, A.; Balin, B.J.; Appelt, D.M. Chlamydia pneumoniae induces Alzheimer-like amyloid plaques in brains of BALB/c mice. Neurobiol. Aging 2004, 25, 419–429. [Google Scholar] [CrossRef] [Green Version]
- Little, C.S.; Joyce, T.A.; Hammond, C.J.; Matta, H.; Cahn, D.; Appelt, D.M.; Balin, B.J. Detection of bacterial antigens and Alzheimer’s disease-like pathology in the central nervous system of BALB/c mice following intranasal infection with a laboratory isolate of Chlamydia pneumoniae. Front. Aging Neurosci. 2014, 6. [Google Scholar] [CrossRef] [Green Version]
- Gieffers, J.; Reusche, E.; Solbach, W.; Maass, M. Failure To Detect Chlamydia pneumoniae in Brain Sections of Alzheimer’s Disease Patients. J. Clin. Microbiol. 2000, 38, 881–882. [Google Scholar] [CrossRef] [Green Version]
- Taylor, G.S.; Vipond, I.B.; Paul, I.D.; Matthews, S.; Wilcock, G.K.; Caul, E.O. Failure to correlate C. pneumoniae with late onset Alzheimer’s disease. Neurology 2002, 59, 142–143. [Google Scholar] [CrossRef]
- Ring, R.H.; Lyons, J.M. Failure To Detect Chlamydia pneumoniae in the Late-Onset Alzheimer’s Brain. J. Clin. Microbiol. 2000, 38, 2591–2594. [Google Scholar] [CrossRef] [Green Version]
- MacIntyre, A.; Abramov, R.; Hammond, C.J.; Hudson, A.P.; Arking, E.J.; Little, C.S.; Appelt, D.M.; Balin, B.J. Chlamydia pneumoniae infection promotes the transmigration of monocytes through human brain endothelial cells. J. Neurosci. Res. 2003, 71, 740–750. [Google Scholar] [CrossRef]
- Zhan, X.; Stamova, B.; Sharp, F.R. Lipopolysaccharide Associates with Amyloid Plaques, Neurons and Oligodendrocytes in Alzheimer’s Disease Brain: A Review. Front. Aging Neurosci. 2018, 10, 42. [Google Scholar] [CrossRef] [Green Version]
- Cunningham, A.; Johnston, S.; Julious, S.; Lampe, F.; Ward, M. Chronic Chlamydia pneumoniae infection and asthma exacerbations in children. Eur. Respir. J. 1998, 11, 345–349. [Google Scholar] [CrossRef] [Green Version]
- Hahn, D.L.; Schure, A.; Patel, K.; Childs, T.; Drizik, E.; Webley, W. Chlamydia pneumoniae-Specific IgE Is Prevalent in Asthma and Is Associated with Disease Severity. PLoS ONE 2012, 7, e35945. [Google Scholar] [CrossRef] [Green Version]
- Iramain, R.; De Jesús, R.; Spitters, C.; Jara, A.; Jimenez, J.; Bogado, N.; Cardozo, L. Chlamydia pneumoniae, and mycoplasma pneumoniae: Are they related to severe asthma in childhood? J. Asthma 2016, 53, 618–621. [Google Scholar] [CrossRef]
- Webley, W.C.; Hahn, D.L. Infection-mediated asthma: Etiology, mechanisms and treatment options, with focus on Chlamydia pneumoniae and macrolides. Respir. Res. 2017, 18, 98. [Google Scholar] [CrossRef] [Green Version]
- Abdulkarim, A.S.; Petrovic, L.M.; Kim, W.R.; Angulo, P.; Lloyd, R.V.; Lindor, K.D. Primary biliary cirrhosis: An infectious disease caused by Chlamydia pneumoniae? J. Hepatol. 2004, 40, 380–384. [Google Scholar] [CrossRef]
- Leung, P.S.C.; Park, O.; Matsumura, S.; Ansari, A.A.; Coppel, R.L.; Gershwin, M.E. Is there a Relation between Chlamydia Infection and Primary Biliary Cirrhosis? Clin. Dev. Immunol. 2003, 10. [Google Scholar] [CrossRef]
- Liu, H.-Y.; Deng, A.-M.; Zhang, J.; Zhou, Y.; Yao, D.-K.; Tu, X.-Q.; Fan, L.-Y.; Zhong, R.-Q. Correlation of Chlamydia pneumoniae infection with primary biliary cirrhosis. World J. Gastroenterol. 2005, 11, 4108–4110. [Google Scholar] [CrossRef]
- Arcos, M.M.; Ruiz, M.F.; Fuentes-Lopez, E.; Alvarez, D.G.; Duarte, d.I.C.; Arrese, M.J.; Riquelme, A.P.; Soza, A.R. Chlamydophila pneumoniae infection is not associated with primary biliary colangitis. Gastroenterol. Latinoam. 2019, 30, 58–63. [Google Scholar]
- Bogdanos, D.P.; Vergani, D. Bacteria and primary biliary cirrhosis. Clin. Rev. Allergy Immunol. 2009, 36, 30–39. [Google Scholar] [CrossRef]
- Davies, B.; Turner, K.M.; Frolund, M.; Ward, H.; May, M.T.; Rasmussen, S.; Benfield, T.; Westh, H.; Danish Chlamydia Study Group. Risk of reproductive complications following chlamydia testing: A population-based retrospective cohort study in Denmark. Lancet Infect. Dis. 2016, 16, 1057–1064. [Google Scholar] [CrossRef] [Green Version]
- Cheong, H.C.; Lee, C.Y.Q.; Cheok, Y.Y.; Tan, G.M.Y.; Looi, C.Y.; Wong, W.F. Chlamydiaceae: Diseases in Primary Hosts and Zoonosis. Microorganisms 2019, 7. [Google Scholar] [CrossRef] [Green Version]
- Storey, G.; Scott, D. Arthritis associated with venereal disease in nineteenth century London. Clin. Rheumatol. 1998, 17, 500–504. [Google Scholar] [CrossRef]
- Taylor-Robinson, D.; Keat, A. How can a causal role for small bacteria in chronic inflammatory arthritides be established or refuted? Ann. Rheum. Dis. 2001, 60, 177. [Google Scholar] [CrossRef] [Green Version]
- Zeidler, H.; Hudson, A.P. Causality of Chlamydiae in arthritis and spondyloarthritis: A plea for increased translational research. Curr. Rheumatol. Rep. 2016, 18, 9. [Google Scholar] [CrossRef]
- Fujita, M.; Hatachi, S.; Yagita, M. Acute Chlamydia pneumoniae infection in the pathogenesis of autoimmune diseases. Lupus 2009, 18, 164–168. [Google Scholar] [CrossRef]
- Gérard, H.C.; Schumacher, H.R.; El-Gabalawy, H.; Goldbach-Mansky, R.; Hudson, A.P. Chlamydia pneumoniae present in the human synovium are viable and metabolically active. Microb. Pathog. 2000, 29, 17–24. [Google Scholar] [CrossRef]
- Moling, O.; Pegoretti, S.; Rielli, M.; Rimenti, G.; Vedovelli, C.; PristerÁ, R.; Mian, P. Chlamydia pneumoniae–Reactive Arthritis and Persistent Infection. Rheumatology 1996, 35, 1189–1190. [Google Scholar] [CrossRef] [Green Version]
- Braun, J.; Laitko, S.; Treharne, J.; Eggens, U.; Wu, P.; Distler, A.; Sieper, J. Chlamydia pneumoniae--a new causative agent of reactive arthritis and undifferentiated oligoarthritis. Ann. Rheum. Dis. 1994, 53, 100–105. [Google Scholar] [CrossRef] [Green Version]
- Schumacher, H.R., Jr.; G’erard, H.e.C.; Arayssi, T.K.; Pando, J.A.; Branigan, P.J.; Saaibi, D.L.; Hudson, A.P. Lower prevalence of Chlamydia pneumoniae DNA compared with Chlamydia trachomatis DNA in synovial tissue of arthritis patients. Arthritis Rheum. 1999, 42, 1889–1893. [Google Scholar] [CrossRef]
- Stavropoulos, P.G.; Soura, E.; Kanelleas, A.; Katsambas, A.; Antoniou, C. Reactive Arthritis. J. Eur. Acad. Dermatol. Venereol. 2015, 29, 415–424. [Google Scholar] [CrossRef]
- Gerard, H.C.; Whittum-Hudson, J.A.; Carter, J.D.; Hudson, A.P. The pathogenic role of Chlamydia in spondyloarthritis. Curr. Opin. Rheumatol. 2010, 22, 363–367. [Google Scholar] [CrossRef] [PubMed]
- Baillet, A.C.; Rehaume, L.M.; Benham, H.; O’Meara, C.P.; Armitage, C.W.; Ruscher, R.; Brizard, G.; Harvie, M.C.G.; Velasco, J.; Hansbro, P.M.; et al. High Chlamydia Burden Promotes Tumor Necrosis Factor–Dependent Reactive Arthritis in SKG Mice. Arthritis Rheumatol. 2015, 67, 1535–1547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Datta, B.; Njau, F.; Thalmann, J.; Haller, H.; Wagner, A.D. Differential infection outcome of Chlamydia trachomatis in human blood monocytes and monocyte-derived dendritic cells. BMC Microbiol. 2014, 14, 209. [Google Scholar] [CrossRef] [PubMed]
- Kebbi-Beghdadi, C.; Cisse, O.; Greub, G. Permissivity of Vero cells, human pneumocytes and human endometrial cells to Waddlia chondrophila. Microbes Infect. 2011, 13, 566–574. [Google Scholar] [CrossRef] [PubMed]
- Nelson, D.E.; Virok, D.P.; Wood, H.; Roshick, C.; Johnson, R.M.; Whitmire, W.M.; Crane, D.D.; Steele-Mortimer, O.; Kari, L.; McClarty, G.; et al. Chlamydial IFN-gamma immune evasion is linked to host infection tropism. Proc. Natl. Acad. Sci. USA 2005, 102, 10658–10663. [Google Scholar] [CrossRef] [Green Version]
- Cheong, H.C.; Lee, C.Y.Q.; Cheok, Y.Y.; Shankar, E.M.; Sabet, N.S.; Tan, G.M.Y.; Movahed, E.; Yeow, T.C.; Sulaiman, S.; Wong, W.F.; et al. CPAF, HSP60 and MOMP antigens elicit pro-inflammatory cytokines production in the peripheral blood mononuclear cells from genital Chlamydia trachomatis-infected patients. Immunobiology 2018. [Google Scholar] [CrossRef]
- Duncan, S.A.; Sahu, R.; Martin, D.; Singh, S.R.; Dennis, V. Modulation of Inflammation Induced by Recombinant MOMP of Chlamydia in Mouse Macrophages by IL-10 is Mediated by Skewing M1 to an M2 Polarizing Macrophage Phenotype. J. Immunol. 2017, 198, 201.23. [Google Scholar]
- Carter, J.D.; Espinoza, L.R.; Inman, R.D.; Sneed, K.B.; Ricca, L.R.; Vasey, F.B.; Valeriano, J.; Stanich, J.A.; Oszust, C.; Gerard, H.C.; et al. Combination antibiotics as a treatment for chronic Chlamydia-induced reactive arthritis: A double-blind, placebo-controlled, prospective trial. Arthritis Rheum. 2010, 62, 1298–1307. [Google Scholar] [CrossRef] [Green Version]
- Carter, J.D.; Valeriano, J.; Vasey, F.B. Doxycycline versus doxycycline and rifampin in undifferentiated spondyloarthropathy, with special reference to chlamydia-induced arthritis. A prospective, randomized 9-month comparison. J. Rheumatol. 2004, 31, 1973–1980. [Google Scholar]
- Carter, J.D. Treating reactive arthritis: Insights for the clinician. Adv. Musculoskelet Dis. 2010, 2, 45–54. [Google Scholar] [CrossRef]
- Rihl, M.; Gu, J.; Baeten, D.; Marker-Hermann, E.; Goodall, J.C.; Gaston, J.S.; Kuipers, J.G.; Zeidler, H.; Yu, D.T. Alpha beta but not gamma delta T cell clones in synovial fluids of patients with reactive arthritis show active transcription of tumour necrosis factor alpha and interferon gamma. Ann. Rheum. Dis. 2004, 63, 1673–1676. [Google Scholar] [CrossRef] [Green Version]
- Meyer, A.; Chatelus, E.; Wendling, D.; Berthelot, J.-M.; Dernis, E.; Houvenagel, E.; Morel, J.; Richer, O.; Schaeverbeke, T.; Gottenberg, J.-E.; et al. Safety and efficacy of anti–tumor necrosis factor α therapy in ten patients with recent-onset refractory reactive arthritis. Arthritis Rheum. 2011, 63, 1274–1280. [Google Scholar] [CrossRef]
- Barber, C.E.; Kim, J.; Inman, R.D.; Esdaile, J.M.; James, M.T. Antibiotics for treatment of reactive arthritis: A systematic review and metaanalysis. J. Rheumatol. 2013, 40, 916–928. [Google Scholar] [CrossRef]
- Sessa, R.; Nicoletti, M.; Di Pietro, M.; Schiavoni, G.; Santino, I.; Zagaglia, C.; Del Piano, M.; Cipriani, P. Chlamydia Pneumoniae and Atherosclerosis: Current State and Future Prospectives. Int. J. Immunopathol. Pharmacol. 2009, 22, 9–14. [Google Scholar] [CrossRef]
- Watson, C.; Alp, N.J. Role of Chlamydia pneumoniae in atherosclerosis. Clin. Sci. 2008, 114, 509. [Google Scholar] [CrossRef]
- Belland, R.J.; Ouellette, S.P.; Gieffers, J.; Byrne, G.I. Chlamydia pneumoniae and atherosclerosis. Cell. Microbiol. 2004, 6, 117–127. [Google Scholar] [CrossRef]
- Saikku, P.; Leinonen, M.; Mattila, K.; Ekman, M.R.; Nieminen, M.S.; Makela, P.H.; Huttunen, J.K.; Valtonen, V. Serological evidence of an association of a novel Chlamydia, TWAR, with chronic coronary heart disease and acute myocardial infarction. Lancet 1988, 2, 983–986. [Google Scholar] [CrossRef]
- Edvinsson, M.; Tallkvist, J.; Nystrom-Rosander, C.; Ilback, N.G. Cholesterol uptake in the mouse aorta increases during Chlamydia pneumoniae infection. Pathog. Dis. 2017, 75. [Google Scholar] [CrossRef]
- Yu, X.-H.; Fu, Y.-C.; Zhang, D.-W.; Yin, K.; Tang, C.-K. Foam cells in atherosclerosis. Clin. Chim. Acta 2013, 424, 245–252. [Google Scholar] [CrossRef] [Green Version]
- Charcot, J.M. De la sclerose laterale amyotrophique. Symptomatologie. Lecons Sur. Les. Mal. Du Syst. Nerv. Faites A La Salpetriere 1880, 2, 227–242. [Google Scholar]
- Marrodan, M.; Alessandro, L.; Farez, M.F.; Correale, J. The role of infections in multiple sclerosis. Mult. Scler. 2019, 25, 891–901. [Google Scholar] [CrossRef]
- Bagos, P.G.; Nikolopoulos, G.; Ioannidis, A. Chlamydia pneumoniae infection and the risk of multiple sclerosis: A meta-analysis. Mult. Scler. J. 2006, 12, 397–411. [Google Scholar] [CrossRef]
- Ikejima, H.; Haranaga, S.; Takemura, H.; Kamo, T.; Takahashi, Y.; Friedman, H.; Yamamoto, Y. PCR-Based Method for Isolation and Detection of Chlamydia pneumoniae DNA in Cerebrospinal Fluids. Clin. Diagn. Lab. Immunol. 2001, 8, 499–502. [Google Scholar] [CrossRef] [Green Version]
- Wilske, B.; Barz, C.; Meinl, E.; Then Bergh, F.; Autenrieth, I.; Wick, M.; Hartmann, M.; Goebels, N.; Hohlfeld, R.; Gürkov, R.; et al. Intrathecal antibody production against Chlamydia pneumoniae in multiple sclerosis is part of a polyspecific immune response. Brain 2001, 124, 1325–1335. [Google Scholar] [CrossRef] [Green Version]
- Godzik, K.L.; O’Brien, E.R.; Wang, S.K.; Kuo, C.C. In vitro susceptibility of human vascular wall cells to infection with Chlamydia pneumoniae. J. Clin. Microbiol. 1995, 33, 2411–2414. [Google Scholar] [CrossRef] [Green Version]
- Gérard, H.C.; Fomicheva, E.; Whittum-Hudson, J.A.; Hudson, A.P. Apolipoprotein E4 enhances attachment of Chlamydophila (Chlamydia) pneumoniae elementary bodies to host cells. Microb. Pathog. 2008, 44, 279–285. [Google Scholar] [CrossRef]
- Stratton, C.W.; Wheldon, D.B. Multiple sclerosis: An infectious syndrome involving Chlamydophila pneumoniae. Trends Microbiol. 2006, 14, 474–479. [Google Scholar] [CrossRef]
- Alonso, A.; Jick, H.; Hernán, M.A.; Jick, S.S. Antibiotic Use and Risk of Multiple Sclerosis. Am. J. Epidemiol. 2006, 163, 997–1002. [Google Scholar] [CrossRef]
- Woessner, R.; Grauer, M.T.; Frese, A.; Bethke, F.; Ginger, T.; Hans, A.; Treib, J. Long-term Antibiotic Treatment with Roxithromycin in Patients with Multiple Sclerosis. Infection 2006, 34, 342. [Google Scholar] [CrossRef]
- 2018 Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2018, 14, 367–429. [CrossRef]
- Ballatore, C.; Lee, V.M.Y.; Trojanowski, J.Q. Tau-mediated neurodegeneration in Alzheimer’s disease and related disorders. Nat. Rev. Neurosci. 2007, 8, 663. [Google Scholar] [CrossRef]
- Ittner, L.M.; Ke, Y.D.; Delerue, F.; Bi, M.; Gladbach, A.; van Eersel, J.; Wölfing, H.; Chieng, B.C.; Christie, M.J.; Napier, I.A.; et al. Dendritic Function of Tau Mediates Amyloid-β Toxicity in Alzheimer’s Disease Mouse Models. Cell 2010, 142, 387–397. [Google Scholar] [CrossRef] [Green Version]
- Boelen, E.; Stassen, F.R.M.; van der Ven, A.J.A.M.; Lemmens, M.A.M.; Steinbusch, H.P.J.; Bruggeman, C.A.; Schmitz, C.; Steinbusch, H.W.M. Detection of amyloid beta aggregates in the brain of BALB/c mice after Chlamydia pneumoniae infection. Acta Neuropathol. 2007, 114, 255–261. [Google Scholar] [CrossRef] [Green Version]
- Gérard, H.C.; Wildt, K.L.; Whittum-Hudson, J.A.; Lai, Z.; Ager, J.; Hudson, A.P. The load of Chlamydia pneumoniae in the Alzheimer’s brain varies with APOE genotype. Microb. Pathog. 2005, 39, 19–26. [Google Scholar] [CrossRef]
- Zhang, R.; Miller, R.G.; Gascon, R.; Champion, S.; Katz, J.; Lancero, M.; Narvaez, A.; Honrada, R.; Ruvalcaba, D.; McGrath, M.S. Circulating endotoxin and systemic immune activation in sporadic amyotrophic lateral sclerosis (sALS). J. Neuroimmunol. 2009, 206, 121–124. [Google Scholar] [CrossRef] [Green Version]
- Pretorius, E.; Bester, J.; Page, M.J.; Kell, D.B. The Potential of LPS-Binding Protein to Reverse Amyloid Formation in Plasma Fibrin of Individuals With Alzheimer-Type Dementia. Front. Aging Neurosci. 2018, 10, 257. [Google Scholar] [CrossRef]
- Zhao, J.; Bi, W.; Xiao, S.; Lan, X.; Cheng, X.; Zhang, J.; Lu, D.; Wei, W.; Wang, Y.; Li, H.; et al. Neuroinflammation induced by lipopolysaccharide causes cognitive impairment in mice. Sci. Rep. 2019, 9, 5790. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.W.; Lee, Y.K.; Yuk, D.Y.; Choi, D.Y.; Ban, S.B.; Oh, K.W.; Hong, J.T. Neuro-inflammation induced by lipopolysaccharide causes cognitive impairment through enhancement of beta-amyloid generation. J. Neuroinflammation 2008, 5, 37. [Google Scholar] [CrossRef] [Green Version]
- Loeb, M.B.; Molloy, D.W.; Smieja, M.; Standish, T.; Goldsmith, C.H.; Mahony, J.; Smith, S.; Borrie, M.; Decoteau, E.; Davidson, W.; et al. A Randomized, Controlled Trial of Doxycycline and Rifampin for Patients with Alzheimer’s Disease. J. Am. Geriatr. Soc. 2004, 52, 381–387. [Google Scholar] [CrossRef]
- Umeda, T.; Tanaka, A.; Sakai, A.; Yamamoto, A.; Sakane, T.; Tomiyama, T. Intranasal rifampicin for Alzheimer’s disease prevention. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2018, 4, 304–313. [Google Scholar] [CrossRef]
- Sakai, A.; Yamashita, M.; Tomiyama, T.; Umeda, T.; Mori, H.; Ono, K.; Yamada, M.; Mizuguchi, M.; Klein, W.L. Rifampicin is a candidate preventive medicine against amyloid-β and tau oligomers. Brain 2016, 139, 1568–1586. [Google Scholar] [CrossRef] [Green Version]
- Tomiyama, T.; Asano, S.; Suwa, Y.; Morita, T.; Kataoka, K.; Mori, H.; Endo, N. Rifampicin Prevents the Aggregation and Neurotoxicity of Amyloid β Protein in Vitro. Biochem. Biophys. Res. Commun. 1994, 204, 76–83. [Google Scholar] [CrossRef]
- Webley, W.C.; Salva, P.S.; Andrzejewski, C.; Cirino, F.; West, C.A.; Tilahun, Y.; Stuart, E.S. The Bronchial Lavage of Pediatric Patients with Asthma Contains Infectious Chlamydia. Am. J. Respir. Crit. Care Med. 2005, 171, 1083–1088. [Google Scholar] [CrossRef]
- Hahn, D.L.; Webley, W. Chronic Chlamydia pneumoniae lung infection: A neglected explanation for macrolide effects in wheezing and asthma? Lancet Respir. Med. 2016, 4, e8. [Google Scholar] [CrossRef]
- Johnston, S.L.; Martin, R.J. Chlamydophila pneumoniae and Mycoplasma pneumoniae. Am. J. Respir. Crit. Care Med. 2005, 172, 1078–1089. [Google Scholar] [CrossRef] [Green Version]
- Von Hertzen, L.C. Role of persistent infection in the control and severity of asthma: Focus on Chlamydia pneumonia. Eur. Respir. J. 2002, 19, 546–556. [Google Scholar] [CrossRef] [Green Version]
- Cook, P.J.; Davies, P.; Tunnicliffe, W.; Ayres, J.G.; Honeybourne, D.; Wise, R. Chlamydia pneumoniae and asthma. Thorax 1998, 53, 254–259. [Google Scholar] [CrossRef] [Green Version]
- Hahn, D.L.; Grasmick, M.; Hetzel, S.; Yale, S. Azithromycin for Bronchial Asthma in Adults: An Effectiveness Trial. J. Am. Board Fam. Med. 2012, 25, 442. [Google Scholar] [CrossRef] [Green Version]
- Tamimi, A.; Serdarevic, D.; Hanania, N.A. The effects of cigarette smoke on airway inflammation in asthma and COPD: Therapeutic implications. Respir. Med. 2012, 106, 319–328. [Google Scholar] [CrossRef] [Green Version]
- Shemer-Avni, Y.; Lieberman, D. Chlamydia pneumoniae-Induced Ciliostasis in Ciliated Bronchial Epithelial Cells. J. Infect. Dis. 1995, 171, 1274–1278. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, M.M.; Gershwin, M.E. Primary Biliary Cirrhosis. New Engl. J. Med. 2005, 353, 1261–1273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neuberger, J.; Thomson, R. PBC and AMA—what is the connection? Hepatology 1999, 29, 271–276. [Google Scholar] [CrossRef]


{kind=link}
| Diseases | Supporting or Contradicting Evidence | Mechanisms of Pathogenesis |
|---|---|---|
| Reactive Arthritis (ReA) | Supporting: Presence of C. trachomatis DNA, antigens, EB in the synovial fluid; elevated serum anti-C. trachomatis antibodies in ReA patients [13,14,15]. |
|
| Atherosclerosis | Supporting: Presence of C. pneumoniae in atherosclerotic plaques exacerbates disease pathology; elevated anti-C. pneumoniae antibodies among patients [23,24,25]. |
|
| Multiple Sclerosis (MS) | Supporting: High percentage of positive C. pneumoniae infection using culture method; positive amplification of chlamydial OmpA gene [30,31]; presence of anti-C. pneumoniae antibodies in MS patients [31,32,33,34]. Contradicting: Failure to detect bacteria in MS patients using PCR or culture methods [35]; presence of bacteria in other neurological diseases in addition to MS [36]. |
|
| Alzheimer’s Disease | Supporting: Presence of live and metabolically active C. pneumoniae in brain of Alzheimer’s disease patients [42,43,44]; intranasal C. pneumoniae-infected mice developed brain Aβ deposits [45,46]. Contradicting: Failure of bacterial detection in patients’ brain section [36,47,48], or PCR amplification [49]. |
|
| Asthma | Supporting: C. pneumoniae-specific IgA, IgE, IgG, and IgM antibodies were detected in asthmatic patients [52,53,54]; IgE showed a positive linear association with asthma severity [53]. Children who reported multiple episodes of asthma were PCR positive for C. pneumoniae [52]. | C. pneumoniae contributes to asthma by causing increased secretion of inflammatory cytokines and chemokines [52,55]. |
| Primary Biliary Cirrhosis (PBC) | Supporting: C. pneumoniae antigens and 16S rRNA was detected in the explanted liver tissue or biopsy [56]; elevated anti-chlamydial antibodies among PBC or anti-mitochondrial antibodies (AMA)-positive PBC patients [57,58]. Contradicting: Absence of bacteria or bacterial DNA in the liver tissues; no significant serum anti-C. pneumoniae IgG in PBC patients [57,58,59]. | Potentially due to molecular mimicry between bacteria and host antigens or direct presence of the bacteria at liver [60], however an exact mechanism is still unknown. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cheok, Y.Y.; Lee, C.Y.Q.; Cheong, H.C.; Looi, C.Y.; Wong, W.F. Chronic Inflammatory Diseases at Secondary Sites Ensuing Urogenital or Pulmonary Chlamydia Infections. Microorganisms 2020, 8, 127. https://doi.org/10.3390/microorganisms8010127
Cheok YY, Lee CYQ, Cheong HC, Looi CY, Wong WF. Chronic Inflammatory Diseases at Secondary Sites Ensuing Urogenital or Pulmonary Chlamydia Infections. Microorganisms. 2020; 8(1):127. https://doi.org/10.3390/microorganisms8010127
Chicago/Turabian StyleCheok, Yi Ying, Chalystha Yie Qin Lee, Heng Choon Cheong, Chung Yeng Looi, and Won Fen Wong. 2020. "Chronic Inflammatory Diseases at Secondary Sites Ensuing Urogenital or Pulmonary Chlamydia Infections" Microorganisms 8, no. 1: 127. https://doi.org/10.3390/microorganisms8010127