Transmission Network of Deer-Borne Mycobacterium bovis Infection Revealed by a WGS Approach

 , , and
, , and
Abstract
:1. Introduction
2. Material and Methods
2.1. Ethical Statement
2.2. Bacterial Strains
2.3. Whole Genome Sequencing and Variant Calling
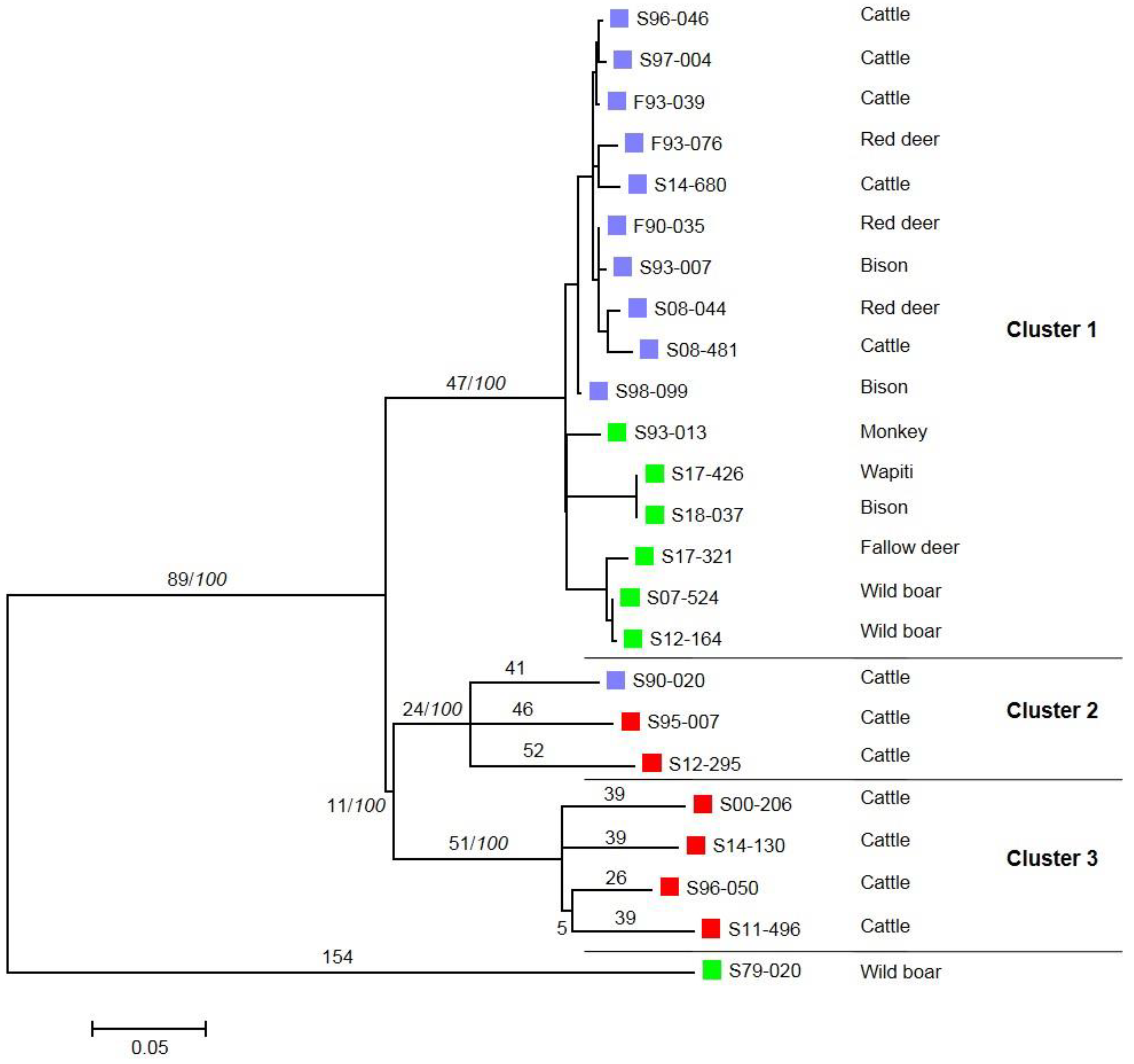
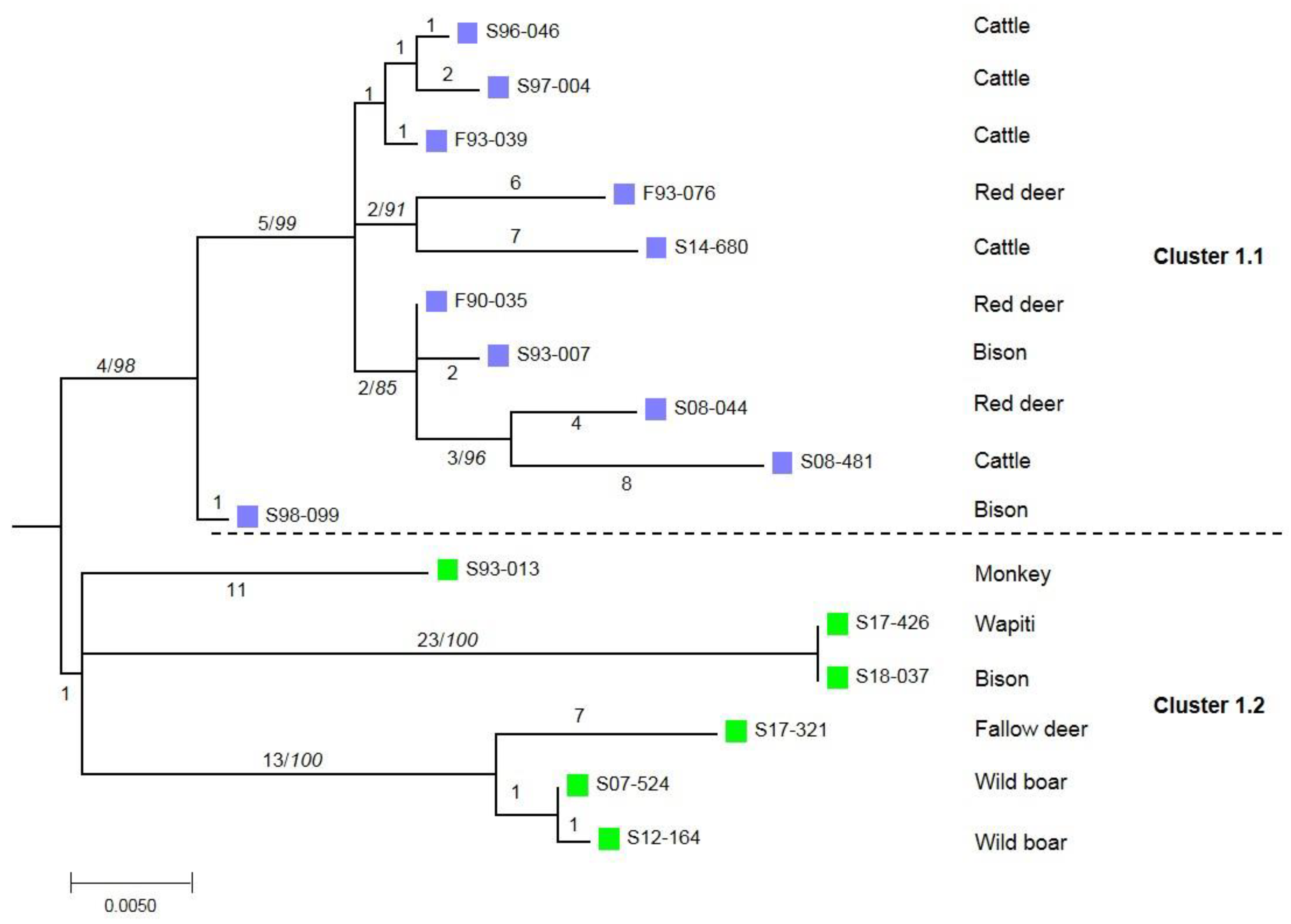
3. Results
4. Discussion and Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Muller, B.; Durr, S.; Alonso, S.; Hattendorf, J.; Laisse, C.J.; Parsons, S.D.; van Helden, P.D.; Zinsstag, J. Zoonotic Mycobacterium bovis-induced tuberculosis in humans. Emerg. Infect. Dis. 2013, 19, 899–908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hauer, A.; De Cruz, K.; Cochard, T.; Godreuil, S.; Karoui, C.; Henault, S.; Bulach, T.; Banuls, A.L.; Biet, F.; Boschiroli, M.L. Genetic evolution of Mycobacterium bovis causing tuberculosis in livestock and wildlife in France since 1978. PLoS ONE 2015, 10, e0117103. [Google Scholar] [CrossRef] [PubMed]
- Gortázar, C.; Delahay, R.J.; McDonald, R.A.; Boadella, M.; Wilson, G.J.; Gavier-Widen, D.; Acevedo, P. The status of tuberculosis in European wild mammals. Mammal Rev. 2012, 42, 193–206. [Google Scholar] [CrossRef]
- Griffin, J.F.; Mackintosh, C.G. Tuberculosis in deer: Perceptions, problems and progress. Vet. J. 2000, 160, 202–219. [Google Scholar] [CrossRef] [PubMed]
- Zanella, G.; Duvauchelle, A.; Hars, J.; Moutou, F.; Boschiroli, M.L.; Durand, B. Patterns of lesions of bovine tuberculosis in wild red deer and wild boar. Vet. Rec. 2008, 163, 43–47. [Google Scholar] [CrossRef] [PubMed]
- Mackintosh, C.G.; de Lisle, G.W.; Collins, D.M.; Griffin, J.F. Mycobacterial diseases of deer. N. Z. Vet. J. 2004, 52, 163–174. [Google Scholar] [CrossRef]
- Martin-Hernando, M.P.; Torres, M.J.; Aznar, J.; Negro, J.J.; Gandia, A.; Gortazar, C. Distribution of lesions in red and fallow deer naturally infected with Mycobacterium bovis. J. Comp. Pathol. 2010, 142, 43–50. [Google Scholar] [CrossRef] [Green Version]
- Miller, M.; Olea-Popelka, F. One Health in the shrinking world: Experiences with tuberculosis at the human-livestock-wildlife interface. Comp. Immunol. Microbiol. Infect. Dis. 2013, 36, 263–268. [Google Scholar] [CrossRef]
- Dalovisio, J.R.; Stetter, M.; Mikota-Wells, S. Rhinoceros’ rhinorrhea: Cause of an outbreak of infection due to airborne Mycobacterium bovis in zookeepers. Clin. Infect. Dis. 1992, 15, 598–600. [Google Scholar] [CrossRef]
- Kiers, A.; Klarenbeek, A.; Mendelts, B.; Van Soolingen, D.; Koeter, G. Transmission of Mycobacterium pinnipedii to humans in a zoo with marine mammals. Int. J. Tuberc. Lung Dis. 2008, 12, 1469–1473. [Google Scholar]
- Montali, R.J.; Mikota, S.K.; Cheng, L.I. Mycobacterium tuberculosis in zoo and wildlife species. Rev. Sci. Tech. 2001, 20, 291–303. [Google Scholar] [CrossRef] [PubMed]
- Murphree, R.; Warkentin, J.V.; Dunn, J.R.; Schaffner, W.; Jones, T.F. Elephant-to-human transmission of tuberculosis, 2009. Emerg. Infect. Dis. 2011, 17, 366–371. [Google Scholar] [CrossRef] [PubMed]
- Gharib Mombeni, E.; Mosavari, N.; Moradi Gravand, M.; Amir Rezai, A.; Keshavarz, R.; Tadayon, K.; Bakhshi, R.; Behmanesh, R. First Report of Mycobacterium bovis Isolation from a European Fallow Deer (Dama Dama Dama) in Iran. Iran. J. Public Health 2016, 45, 814–816. [Google Scholar] [PubMed]
- Zhang, J.; Abadia, E.; Refregier, G.; Tafaj, S.; Boschiroli, M.L.; Guillard, B.; Andremont, A.; Ruimy, R.; Sola, C. Mycobacterium tuberculosis complex CRISPR genotyping: Improving efficiency, throughput and discriminative power of ‘spoligotyping’ with new spacers and a microbead-based hybridization assay. J. Med. Microbiol. 2010, 59, 285–294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skuce, R.A.; McDowell, S.W.; Mallon, T.R.; Luke, B.; Breadon, E.L.; Lagan, P.L.; McCormick, C.M.; McBride, S.H.; Pollock, J.M. Discrimination of isolates of Mycobacterium bovis in Northern Ireland on the basis of variable numbers of tandem repeats (VNTRs). Vet. Rec. 2005, 157, 501–504. [Google Scholar] [CrossRef]
- Hauer, A.; Michelet, L.; De Cruz, K.; Cochard, T.; Branger, M.; Karoui, C.; Henault, S.; Biet, F.; Boschiroli, M.L. MIRU-VNTR allelic variability depends on Mycobacterium bovis clonal group identity. Infect. Genet. Evol. 2016, 45, 165–169. [Google Scholar] [CrossRef]
- Boschiroli, M.L.; Michelet, L.; Hauer, A.; De Cruz, K.; Courcoul, A.; Hénault, S.; Palisson, A.; Karoui, C.; Biet, F.; Zanella, G. Tuberculose bovine en France: Cartographie des souches de Mycobacterium bovis entre 2000–2013. Bull. Epid. St. Anim. Alim. 2015, 70, 2–8. [Google Scholar]
- Cavalerie, L.; Courcoul, A.; Boschiroli, M.L.; Réveillaud, E.; Gay, P. Bovine Tuberculosis in France in 2014: A stable situation. Bull. Epid. St. Anim. Alim. 2015, 71, 4–11. [Google Scholar]
- Hauer, A.; Michelet, L.; Cochard, T.; Branger, M.; Nunez, J.; Boschiroli, M.L.; Biet, F. Accurate Phylogenetic Relationships Among Mycobacterium bovis Strains Circulating in France Based on Whole Genome Sequencing and Single Nucleotide Polymorphism Analysis. Front. Microbiol. 2019, 10, 955. [Google Scholar] [CrossRef] [Green Version]
- Turankar, R.P.; Lavania, M.; Chaitanya, V.S.; Sengupta, U.; Darlong, J.; Darlong, F.; Siva Sai, K.S.; Jadhav, R.S. Single nucleotide polymorphism-based molecular typing of M. leprae from multicase families of leprosy patients and their surroundings to understand the transmission of leprosy. Clin. Microbiol. Infect. 2014, 20, O142–O149. [Google Scholar] [CrossRef] [Green Version]
- Roetzer, A.; Diel, R.; Kohl, T.A.; Ruckert, C.; Nubel, U.; Blom, J.; Wirth, T.; Jaenicke, S.; Schuback, S.; Rusch-Gerdes, S.; et al. Whole genome sequencing versus traditional genotyping for investigation of a Mycobacterium tuberculosis outbreak: A longitudinal molecular epidemiological study. PLoS Med. 2013, 10, e1001387. [Google Scholar] [CrossRef] [PubMed]
- Biek, R.; O’Hare, A.; Wright, D.; Mallon, T.; McCormick, C.; Orton, R.J.; McDowell, S.; Trewby, H.; Skuce, R.A.; Kao, R.R. Whole genome sequencing reveals local transmission patterns of Mycobacterium bovis in sympatric cattle and badger populations. PLoS Pathog. 2012, 8, e1003008. [Google Scholar] [CrossRef] [PubMed]
- Trewby, H.; Wright, D.; Breadon, E.L.; Lycett, S.J.; Mallon, T.R.; McCormick, C.; Johnson, P.; Orton, R.J.; Allen, A.R.; Galbraith, J.; et al. Use of bacterial whole-genome sequencing to investigate local persistence and spread in bovine tuberculosis. Epidemics 2016, 14, 26–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henault, S.; Karoui, C.; Boschiroli, M.L. A PCR-based method for tuberculosis detection in wildlife. Dev. Biol. 2006, 126, 23–132, discussion 325–126. [Google Scholar]
- Roring, S.; Scott, A.; Brittain, D.; Walker, I.; Hewinson, G.; Neill, S.; Skuce, R. Development of variable-number tandem repeat typing of Mycobacterium bovis: Comparison of results with those obtained by using existing exact tandem repeats and spoligotyping. J. Clin. Microbiol. 2002, 40, 2126–2133. [Google Scholar] [CrossRef] [Green Version]
- Supply, P.; Allix, C.; Lesjean, S.; Cardoso-Oelemann, M.; Rusch-Gerdes, S.; Willery, E.; Savine, E.; de Haas, P.; van Deutekom, H.; Roring, S.; et al. Proposal for standardization of optimized mycobacterial interspersed repetitive unit-variable-number tandem repeat typing of Mycobacterium tuberculosis. J. Clin. Microbiol. 2006, 44, 4498–4510. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [Green Version]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. jModelTest 2: More models, new heuristics and parallel computing. Nat. Methods 2012, 9, 772. [Google Scholar] [CrossRef] [Green Version]
- Richomme, C.; Rivière, J.; Hars, J.; Boschiroli, M.L.; Gueneau, E.; Fediaevsky, A.; Dufour, H. Tuberculose bovine: Infection de sangliers dans un parc de chasse. Bull. Epid. St. Anim. Alim. 2013, 56, 14–16. [Google Scholar]
- Thomas, J.; Infantes-Lorenzo, J.A.; Moreno, I.; Romero, B.; Garrido, J.M.; Juste, R.; Dominguez, M.; Dominguez, L.; Gortazar, C.; Risalde, M.A. A new test to detect antibodies against Mycobacterium tuberculosis complex in red deer serum. Vet. J. 2019, 244, 98–103. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| ID | Species | Year of Isolation | Context | Spoligotype | VNTR Profile | Accession Number | ||
|---|---|---|---|---|---|---|---|---|
| 1 | 1979-02442 | S79-020 | Wild boar | 1979 | Park | GB54/SB0121 | 6 3 5 3 11 1 5 7 | ERS3860435 |
| 2 | 1990-08827 | F90-035 | Red deer | 1990 | French herd | GB54/SB0121 | 6 4 5 3 11 2 5 7 | ERS3860436 |
| 3 | 1990-09372 | S90-020 | Cattle | 1990 | French herd | F70/SB0295 | 6 4 5 3 11 2 5 7 | ERS3860437 |
| 4 | 1993-04379 | F93-039 | Cattle | 1993 | French herd | GB54/SB0121 | 6 4 5 3 11 2 5 7 | ERS3860438 |
| 5 | 1993-06259 | S93-007 | Bison | 1993 | French herd | GB54/SB0121 | 6 4 5 3 11 2 5 7 | ERS3860439 |
| 6 | 1993-11575 | F93-076 | Red deer | 1993 | French herd | GB54/SB0121 | 6 4 5 3 11 2 5 7 | ERS3860440 |
| 7 | 1993-11583 | S93-013 | Monkey | 1993 | Park | GB54/SB0121 | 6 4 5 3 11 2 5 7 | ERS3860441 |
| 8 | 1995-00583 | S95-007 | Cattle | 1995 | Foreign-born animal | F070/SB0295 | 6 4 5 3 11 2 5 7 | ERS3860442 |
| 9 | 1996-07003 | S96-046 | Cattle | 1996 | French herd | GB54/SB0121 | 6 4 5 3 11 2 5 7 | ERS3860443 |
| 10 | 1996-08822 | S96-050 | Cattle | 1996 | Foreign-born animal | GB54/SB0121 | 6 4 5 3 11 2 5 7 | ERS3860444 |
| 11 | 1997-00766 | S97-004 | Cattle | 1997 | French herd | GB54/SB0121 | 6 4 5 3 11 2 5 7 | ERS3860445 |
| 12 | 1998-12646 | S98-099 | Bison | 1998 | French herd | GB54/SB0121 | 6 4 5 3 11 2 5 7 | ERS3860446 |
| 13 | 00-06572 | S00-206 | Cattle | 2000 | Foreign-born animal | GB54/SB0121 | 6 4 5 3 11 2 5 7 | ERS3860447 |
| 14 | 07-03151 | S07-524 | Wild boar | 2007 | Park | GB54/SB0121 | 6 4 5 3 11 2 5 7 | ERS3860448 |
| 15 | 08-00202 | S08-044 | Red deer | 2008 | French herd | GB54/SB0121 | 6 4 5 3 11 2 5 7 | ERS3860449 |
| 16 | 08-02744 | S08-481 | Cattle | 2008 | French herd | GB54/SB0121 | 6 4 5 3 11 2 5 7 | ERS3860450 |
| 17 | D11-02906 | S11-496 | Cattle | 2011 | Foreign-born animal | GB54/SB0121 | 6 4 5 3 11 2 5 7 | ERS3860451 |
| 18 | D12-01540 | S12-164 | Wild boar | 2012 | Park | GB54/SB0121 | 6 4 5 3 11 2 5 7 | ERS3860452 |
| 19 | D12-02012 | S12-295 | Cattle | 2012 | Foreign-born animal | F070/SB0295 | 6 4 5 3 11 2 5 7 | ERS3860453 |
| 20 | D14-00543 | S14-130 | Cattle | 2014 | Foreign-born animal | GB54/SB0121 | 6 4 5 3 11 2 5 7 | ERS3860454 |
| 21 | D14-02802 | S14-680 | Cattle | 2014 | French herd | GB54/SB0121 | 6 4 5 3 11 2 5 7 | ERS3860455 |
| 22 | D17-02544 | S17-321 | Fallow deer | 2017 | Park | GB54/SB0121 | 6 4 5 3 11 2 5 7 | ERS3860456 |
| 23 | D17-03607 | S17-426 | Wapiti | 2017 | Park | GB54/SB0121 | 6 4 5 3 11 2 5 7 | ERS3860457 |
| 24 | D18-00301 | S18-037 | Bison | 2018 | Park | GB54/SB0121 | 6 4 5 3 11 2 5 7 | ERS3860458 |
| F90-035 | F93-039 | S93-007 | F93-076 | S96-046 | S97-004 | S98-099 | S08-044 | S08-481 | S14-680 | S93-013 | S07-524 | S12-164 | S17-321 | S17-426 | S18-037 | S90-020 | S95-007 | S12-295 | S96-050 | S00-206 | S11-496 | S14-130 | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Cluster 1.1 | F90-035 | |||||||||||||||||||||||
| Cluster 1.1 | F93-039 | 4 | ||||||||||||||||||||||
| Cluster 1.1 | S93-007 | 2 | 6 | |||||||||||||||||||||
| Cluster 1.1 | F93-076 | 10 | 10 | 12 | ||||||||||||||||||||
| Cluster 1.1 | S96-046 | 5 | 3 | 7 | 11 | |||||||||||||||||||
| Cluster 1.1 | S97-004 | 6 | 4 | 8 | 12 | 3 | ||||||||||||||||||
| Cluster 1.1 | S98-099 | 8 | 8 | 10 | 14 | 9 | 10 | |||||||||||||||||
| Cluster 1.1 | S08-044 | 7 | 11 | 9 | 17 | 12 | 13 | 15 | ||||||||||||||||
| Cluster 1.1 | S08-481 | 11 | 15 | 13 | 21 | 16 | 17 | 19 | 12 | |||||||||||||||
| Cluster 1.1 | S14-680 | 11 | 11 | 13 | 13 | 12 | 13 | 15 | 18 | 22 | ||||||||||||||
| Cluster 1.2 | S93-013 | 23 | 23 | 25 | 29 | 24 | 25 | 17 | 30 | 34 | 30 | |||||||||||||
| Cluster 1.2 | S07-524 | 27 | 27 | 29 | 33 | 28 | 29 | 21 | 34 | 38 | 34 | 26 | ||||||||||||
| Cluster 1.2 | S12-164 | 28 | 28 | 30 | 34 | 29 | 30 | 22 | 35 | 39 | 35 | 27 | 1 | |||||||||||
| Cluster 1.2 | S17-321 | 32 | 32 | 34 | 38 | 33 | 34 | 26 | 39 | 43 | 39 | 31 | 9 | 10 | ||||||||||
| Cluster 1.2 | S17-426 | 35 | 35 | 37 | 41 | 36 | 37 | 29 | 42 | 46 | 42 | 34 | 38 | 39 | 43 | |||||||||
| Cluster 1.2 | S18-037 | 35 | 35 | 37 | 41 | 36 | 37 | 29 | 42 | 46 | 42 | 34 | 38 | 39 | 43 | 0 | ||||||||
| Cluster 2 | S90-020 | 134 | 134 | 136 | 140 | 135 | 136 | 128 | 141 | 145 | 141 | 135 | 139 | 140 | 144 | 147 | 147 | |||||||
| Cluster 2 | S95-007 | 139 | 139 | 141 | 145 | 140 | 141 | 133 | 146 | 150 | 146 | 140 | 144 | 145 | 149 | 152 | 152 | 87 | ||||||
| Cluster 2 | S12-295 | 145 | 145 | 147 | 151 | 146 | 147 | 139 | 152 | 156 | 152 | 146 | 150 | 151 | 155 | 158 | 158 | 93 | 98 | |||||
| Cluster 3 | S96-050 | 151 | 151 | 153 | 157 | 152 | 153 | 145 | 158 | 162 | 158 | 152 | 156 | 157 | 161 | 164 | 164 | 147 | 152 | 158 | ||||
| Cluster 3 | S00-206 | 159 | 159 | 161 | 165 | 160 | 161 | 153 | 166 | 170 | 166 | 160 | 164 | 165 | 169 | 172 | 172 | 155 | 160 | 166 | 70 | |||
| Cluster 3 | S11-496 | 164 | 164 | 166 | 170 | 165 | 166 | 158 | 171 | 175 | 171 | 165 | 169 | 170 | 174 | 177 | 177 | 160 | 165 | 171 | 65 | 83 | ||
| Cluster 3 | S14-130 | 159 | 159 | 161 | 165 | 160 | 161 | 153 | 166 | 170 | 166 | 160 | 164 | 165 | 169 | 172 | 172 | 155 | 160 | 166 | 68 | 78 | 81 | |
| Outgroup | S79-020 | 301 | 301 | 303 | 307 | 302 | 303 | 295 | 308 | 312 | 308 | 302 | 306 | 307 | 311 | 314 | 314 | 319 | 324 | 330 | 336 | 344 | 349 | 344 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Michelet, L.; Conde, C.; Branger, M.; Cochard, T.; Biet, F.; Boschiroli, M.L. Transmission Network of Deer-Borne Mycobacterium bovis Infection Revealed by a WGS Approach. Microorganisms 2019, 7, 687. https://doi.org/10.3390/microorganisms7120687
Michelet L, Conde C, Branger M, Cochard T, Biet F, Boschiroli ML. Transmission Network of Deer-Borne Mycobacterium bovis Infection Revealed by a WGS Approach. Microorganisms. 2019; 7(12):687. https://doi.org/10.3390/microorganisms7120687
Chicago/Turabian StyleMichelet, Lorraine, Cyril Conde, Maxime Branger, Thierry Cochard, Franck Biet, and Maria Laura Boschiroli. 2019. "Transmission Network of Deer-Borne Mycobacterium bovis Infection Revealed by a WGS Approach" Microorganisms 7, no. 12: 687. https://doi.org/10.3390/microorganisms7120687