

Gut Microbiota and Respiratory Infections: Insights from Mendelian Randomization
 , ,
, ,
Abstract
:1. Introduction
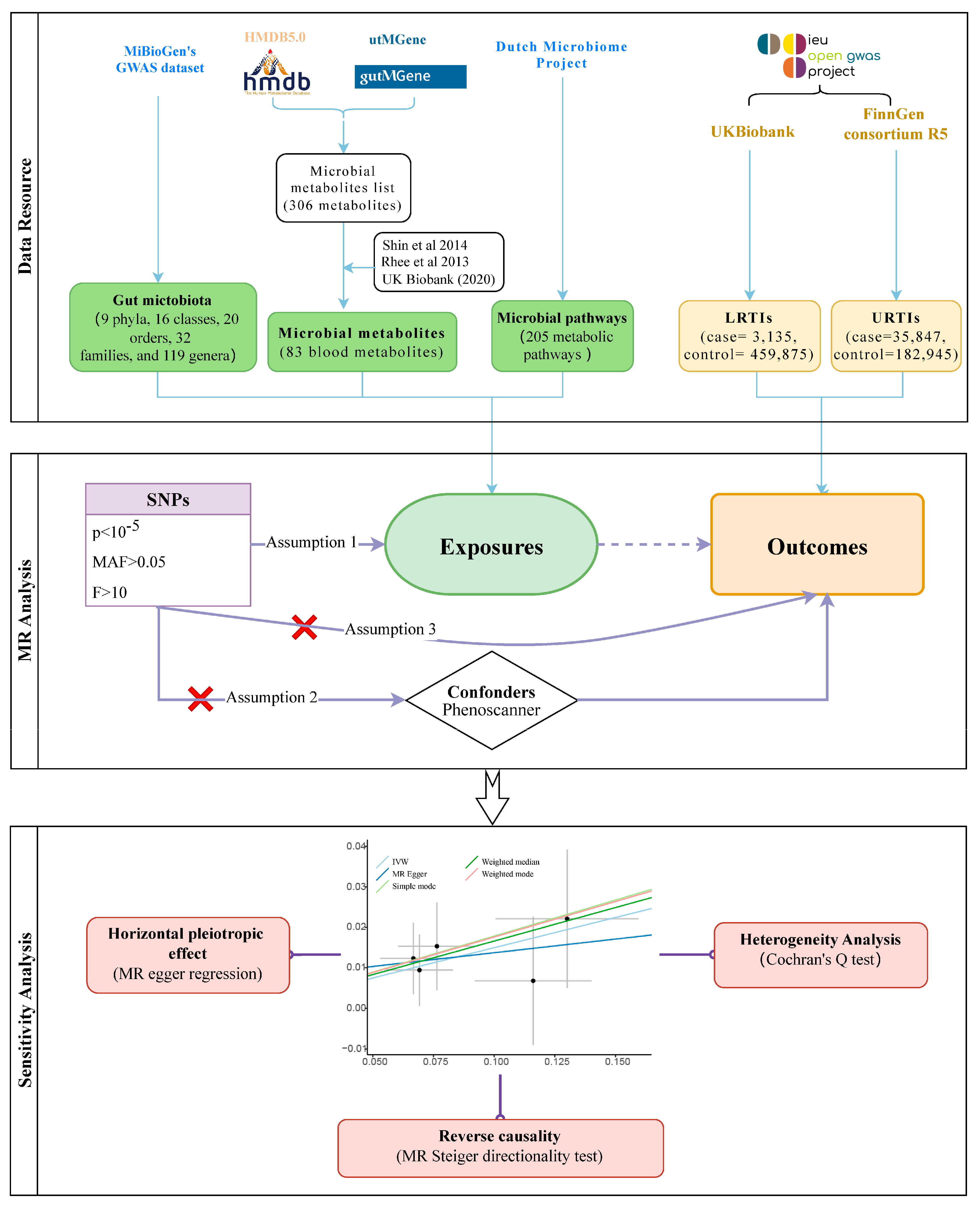
2. Materials and Methods
2.1. Source of Data on Exposure and Outcome
2.2. MR Analysis
2.3. Sensitivity Analysis and Reverse Causation
3. Results
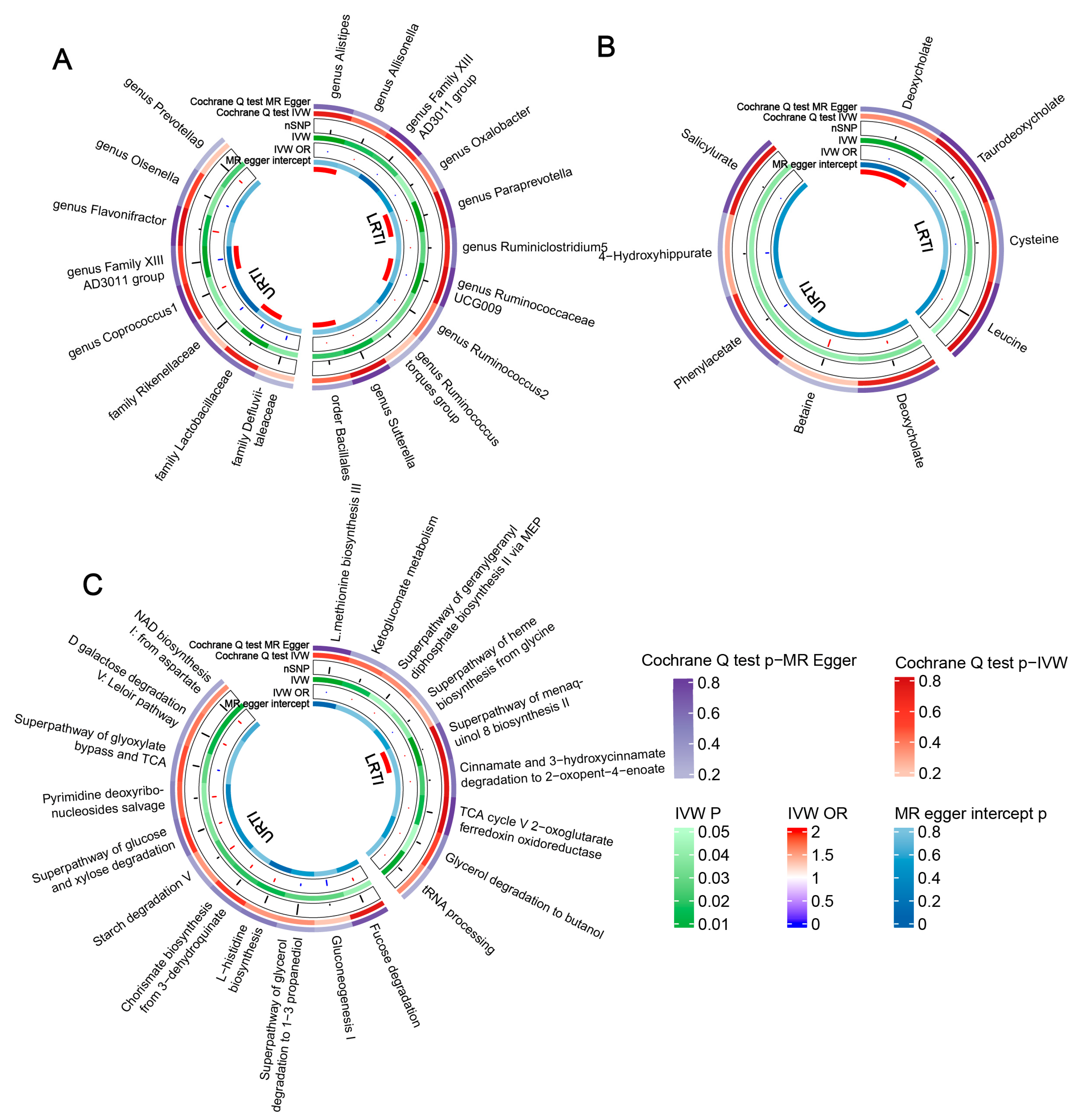
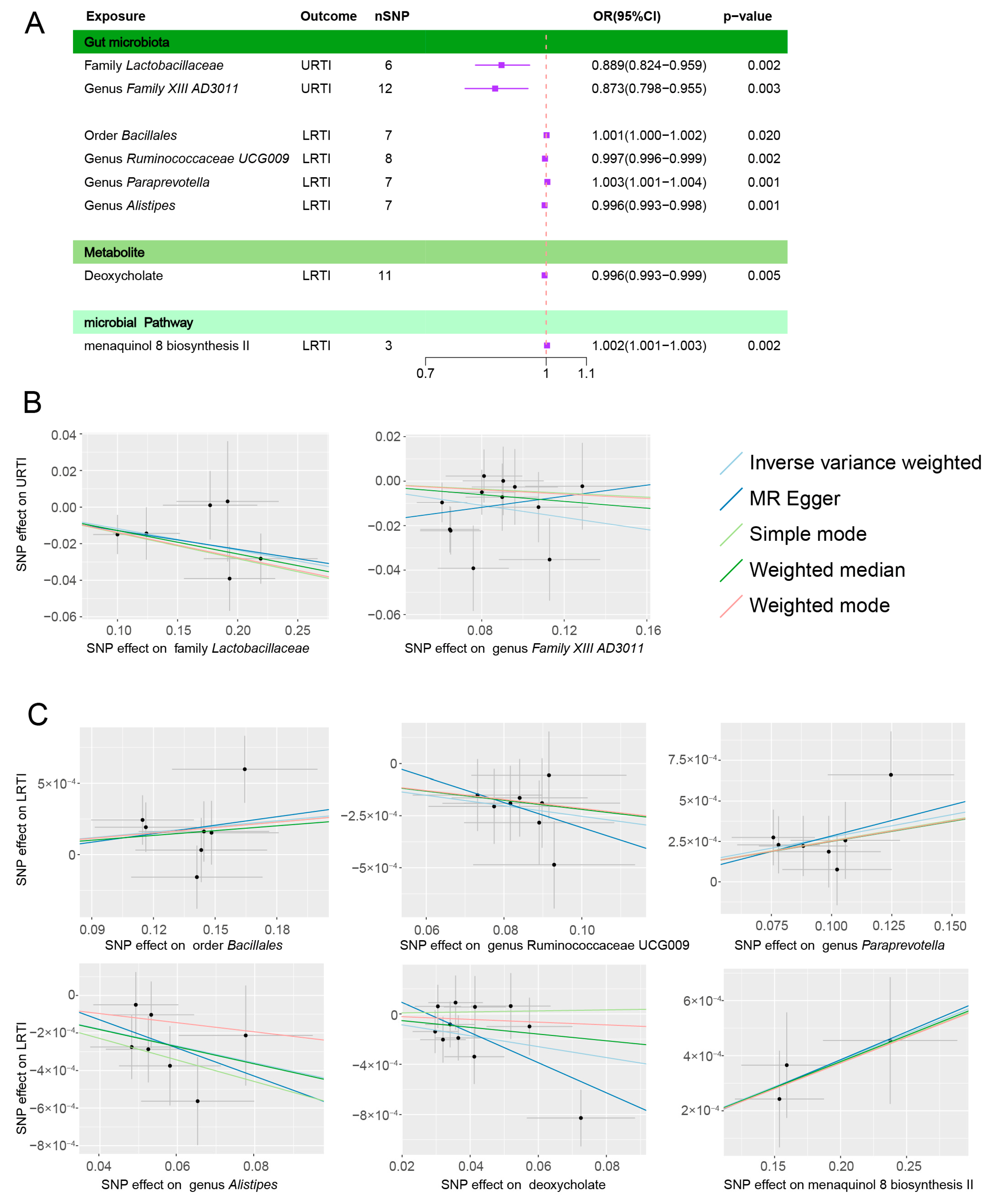
3.1. Gut Microbiota Abundance and Infection
3.1.1. URTIs
3.1.2. LRTIs
3.2. Microbial Metabolites, Pathways, and Infections
3.3. Heterogeneity Test and Reverse Causality
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Priadko, K.; Romano, L.; Olivieri, S.; Romeo, M.; Barone, B.; Sciorio, C.; Spirito, L.; Morelli, M.; Crocetto, F.; Arcaniolo, D.; et al. Intestinal microbiota, intestinal permeability and the urogenital tract: Is there a pathophysiological link? J. Physiol. Pharmacol. 2022, 73. [Google Scholar] [CrossRef]
- Zmora, N.; Suez, J.; Elinav, E. You are what you eat: Diet, health and the gut microbiota. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 35–56. [Google Scholar] [CrossRef] [PubMed]
- Adak, A.; Khan, M.R. An insight into gut microbiota and its functionalities. Cell. Mol. Life Sci. 2019, 76, 473–493. [Google Scholar] [CrossRef]
- Weersma, R.K.; Zhernakova, A.; Fu, J. Interaction between drugs and the gut microbiome. Gut 2020, 69, 1510–1519. [Google Scholar] [CrossRef] [PubMed]
- Becattini, S.; Taur, Y.; Pamer, E.G. Antibiotic-Induced Changes in the Intestinal Microbiota and Disease. Trends Mol. Med. 2016, 22, 458–478. [Google Scholar] [CrossRef]
- Shimizu, K.; Ojima, M.; Ogura, H. Gut Microbiota and Probiotics/Synbiotics for Modulation of Immunity in Critically Ill Patients. Nutrients 2021, 13, 2439. [Google Scholar] [CrossRef]
- Zama, D.; Totaro, C.; Biscardi, L.; Rocca, A.; Turroni, S.; Brigidi, P.; Lanari, M. The Relationship between Gut Microbiota and Respiratory Tract Infections in Childhood: A Narrative Review. Nutrients 2022, 14, 2992. [Google Scholar] [CrossRef]
- Sundararaman, A.; Ray, M.; Ravindra, P.V.; Halami, P.M. Role of probiotics to combat viral infections with emphasis on COVID-19. Appl. Microbiol. Biotechnol. 2020, 104, 8089–8104. [Google Scholar] [CrossRef]
- Zhao, Y.; Dong, B.R.; Hao, Q. Probiotics for preventing acute upper respiratory tract infections. Cochrane Database Syst. Rev. 2022, 8, Cd006895. [Google Scholar] [CrossRef]
- Bo, L.; Li, J.; Tao, T.; Bai, Y.; Ye, X.; Hotchkiss, R.S.; Kollef, M.H.; Crooks, N.H.; Deng, X. Probiotics for preventing ventilator-associated pneumonia. Cochrane Database Syst. Rev. 2014, 10, Cd009066. [Google Scholar] [CrossRef]
- Chunxi, L.; Haiyue, L.; Yanxia, L.; Jianbing, P.; Jin, S. The Gut Microbiota and Respiratory Diseases: New Evidence. J. Immunol. Res. 2020, 2020, 2340670. [Google Scholar] [CrossRef] [PubMed]
- Thibeault, C.; Suttorp, N.; Opitz, B. The microbiota in pneumonia: From protection to predisposition. Sci. Transl. Med. 2021, 13, eaba0501. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Li, M.; Gao, Y.; Liu, M.; Shi, S.; Shi, J.; Yang, K.; Zhou, Z.; Tian, J. Using Mendelian randomization as the cornerstone for causal inference in epidemiology. Environ. Sci. Pollut. Res. Int. 2022, 29, 5827–5839. [Google Scholar] [CrossRef] [PubMed]
- Benn, M.; Nordestgaard, B.G. From genome-wide association studies to Mendelian randomization: Novel opportunities for understanding cardiovascular disease causality, pathogenesis, prevention, and treatment. Cardiovasc. Res. 2018, 114, 1192–1208. [Google Scholar] [CrossRef]
- Ference, B.A.; Holmes, M.V.; Smith, G.D. Using Mendelian Randomization to Improve the Design of Randomized Trials. Cold Spring Harb. Perspect. Med. 2021, 11, a040980. [Google Scholar] [CrossRef]
- Skrivankova, V.W.; Richmond, R.C.; Woolf, B.A.R.; Yarmolinsky, J.; Davies, N.M.; Swanson, S.A.; VanderWeele, T.J.; Higgins, J.P.T.; Timpson, N.J.; Dimou, N.; et al. Strengthening the Reporting of Observational Studies in Epidemiology Using Mendelian Randomization: The STROBE-MR Statement. JAMA 2021, 326, 1614–1621. [Google Scholar] [CrossRef]
- Kurilshikov, A.; Medina-Gomez, C.; Bacigalupe, R.; Radjabzadeh, D.; Wang, J.; Demirkan, A.; Le Roy, C.I.; Raygoza Garay, J.A.; Finnicum, C.T.; Liu, X.; et al. Large-scale association analyses identify host factors influencing human gut microbiome composition. Nat. Genet. 2021, 53, 156–165. [Google Scholar] [CrossRef]
- Wishart, D.S.; Guo, A.; Oler, E.; Wang, F.; Anjum, A.; Peters, H.; Dizon, R.; Sayeeda, Z.; Tian, S.; Lee, B.L.; et al. HMDB 5.0: The Human Metabolome Database for 2022. Nucleic Acids Res. 2022, 50, D622–D631. [Google Scholar] [CrossRef]
- Cheng, L.; Qi, C.; Zhuang, H.; Fu, T.; Zhang, X. gutMDisorder: A comprehensive database for dysbiosis of the gut microbiota in disorders and interventions. Nucleic Acids Res. 2020, 48, D554–D560. [Google Scholar] [CrossRef]
- Shin, S.Y.; Fauman, E.B.; Petersen, A.K.; Krumsiek, J.; Santos, R.; Huang, J.; Arnold, M.; Erte, I.; Forgetta, V.; Yang, T.P.; et al. An atlas of genetic influences on human blood metabolites. Nat. Genet. 2014, 46, 543–550. [Google Scholar] [CrossRef]
- Rhee, E.P.; Ho, J.E.; Chen, M.H.; Shen, D.; Cheng, S.; Larson, M.G.; Ghorbani, A.; Shi, X.; Helenius, I.T.; O’Donnell, C.J.; et al. A genome-wide association study of the human metabolome in a community-based cohort. Cell Metab. 2013, 18, 130–143. [Google Scholar] [CrossRef]
- Lopera-Maya, E.A.; Kurilshikov, A.; van der Graaf, A.; Hu, S.; Andreu-Sánchez, S.; Chen, L.; Vila, A.V.; Gacesa, R.; Sinha, T.; Collij, V.; et al. Effect of host genetics on the gut microbiome in 7,738 participants of the Dutch Microbiome Project. Nat. Genet. 2022, 54, 143–151. [Google Scholar] [CrossRef]
- Hemani, G.; Zheng, J.; Elsworth, B.; Wade, K.H.; Haberland, V.; Baird, D.; Laurin, C.; Burgess, S.; Bowden, J.; Langdon, R.; et al. The MR-Base platform supports systematic causal inference across the human phenome. Elife 2018, 7, e34408. [Google Scholar] [CrossRef]
- Ouyang, Y.; Chen, Y.; Wang, G.; Song, Y.; Zhao, H.; Xiao, B.; Yang, Z.; Long, L. Genetically proxied gut microbiota, gut metabolites with risk of epilepsy and the subtypes: A bi-directional Mendelian randomization study. Front. Mol. Neurosci. 2022, 15, 994270. [Google Scholar] [CrossRef]
- Burgess, S.; Thompson, S.G. Bias in causal estimates from Mendelian randomization studies with weak instruments. Stat. Med. 2011, 30, 1312–1323. [Google Scholar] [CrossRef]
- Li, P.; Wang, H.; Guo, L.; Gou, X.; Chen, G.; Lin, D.; Fan, D.; Guo, X.; Liu, Z. Association between gut microbiota and preeclampsia-eclampsia: A two-sample Mendelian randomization study. BMC Med. 2022, 20, 443. [Google Scholar] [CrossRef]
- Ning, J.; Huang, S.Y.; Chen, S.D.; Zhang, Y.R.; Huang, Y.Y.; Yu, J.T. Investigating Casual Associations Among Gut Microbiota, Metabolites, and Neurodegenerative Diseases: A Mendelian Randomization Study. J. Alzheimers Dis. 2022, 87, 211–222. [Google Scholar] [CrossRef]
- Staley, J.R.; Blackshaw, J.; Kamat, M.A.; Ellis, S.; Surendran, P.; Sun, B.B.; Paul, D.S.; Freitag, D.; Burgess, S.; Danesh, J.; et al. PhenoScanner: A database of human genotype-phenotype associations. Bioinformatics 2016, 32, 3207–3209. [Google Scholar] [CrossRef]
- Burgess, S.; Thompson, S.G. Interpreting findings from Mendelian randomization using the MR-Egger method. Eur. J. Epidemiol. 2017, 32, 377–389. [Google Scholar] [CrossRef]
- Bowden, J.; Del Greco, M.F.; Minelli, C.; Zhao, Q.; Lawlor, D.A.; Sheehan, N.A.; Thompson, J.; Davey Smith, G. Improving the accuracy of two-sample summary-data Mendelian randomization: Moving beyond the NOME assumption. Int. J. Epidemiol. 2019, 48, 728–742. [Google Scholar] [CrossRef]
- Bowden, J.; Davey Smith, G.; Burgess, S. Mendelian randomization with invalid instruments: Effect estimation and bias detection through Egger regression. Int. J. Epidemiol. 2015, 44, 512–525. [Google Scholar] [CrossRef]
- Hemani, G.; Tilling, K.; Davey Smith, G. Orienting the causal relationship between imprecisely measured traits using GWAS summary data. PLoS Genet. 2017, 13, e1007081. [Google Scholar] [CrossRef]
- Li, N.; Wang, Y.; Wei, P.; Min, Y.; Yu, M.; Zhou, G.; Yuan, G.; Sun, J.; Dai, H.; Zhou, E.; et al. Causal Effects of Specific Gut Microbiota on Chronic Kidney Diseases and Renal Function-A Two-Sample Mendelian Randomization Study. Nutrients 2023, 15, 360. [Google Scholar] [CrossRef]
- Kamat, M.A.; Blackshaw, J.A.; Young, R.; Surendran, P.; Burgess, S.; Danesh, J.; Butterworth, A.S.; Staley, J.R. PhenoScanner V2: An expanded tool for searching human genotype-phenotype associations. Bioinformatics 2019, 35, 4851–4853. [Google Scholar] [CrossRef]
- Eribo, O.A.; du Plessis, N.; Chegou, N.N. The Intestinal Commensal, Bacteroides fragilis, Modulates Host Responses to Viral Infection and Therapy: Lessons for Exploration during Mycobacterium tuberculosis Infection. Infect. Immun. 2022, 90, e0032121. [Google Scholar] [CrossRef]
- Ichinohe, T.; Pang, I.K.; Kumamoto, Y.; Peaper, D.R.; Ho, J.H.; Murray, T.S.; Iwasaki, A. Microbiota regulates immune defense against respiratory tract influenza A virus infection. Proc. Natl. Acad. Sci. USA 2011, 108, 5354–5359. [Google Scholar] [CrossRef]
- Chen, J.; Rao, J.N.; Zou, T.; Liu, L.; Marasa, B.S.; Xiao, L.; Zeng, X.; Turner, D.J.; Wang, J.Y. Polyamines are required for expression of Toll-like receptor 2 modulating intestinal epithelial barrier integrity. Am. J. Physiol. Gastrointest. Liver Physiol. 2007, 293, G568–G576. [Google Scholar] [CrossRef]
- Ghosh, S.; Whitley, C.S.; Haribabu, B.; Jala, V.R. Regulation of Intestinal Barrier Function by Microbial Metabolites. Cell. Mol. Gastroenterol. Hepatol. 2021, 11, 1463–1482. [Google Scholar] [CrossRef]
- Lawley, T.D.; Walker, A.W. Intestinal colonization resistance. Immunology 2013, 138, 1–11. [Google Scholar] [CrossRef]
- Fukuda, S.; Toh, H.; Hase, K.; Oshima, K.; Nakanishi, Y.; Yoshimura, K.; Tobe, T.; Clarke, J.M.; Topping, D.L.; Suzuki, T.; et al. Bifidobacteria can protect from enteropathogenic infection through production of acetate. Nature 2011, 469, 543–547. [Google Scholar] [CrossRef]
- Berg, R.D.; Garlington, A.W. Translocation of certain indigenous bacteria from the gastrointestinal tract to the mesenteric lymph nodes and other organs in a gnotobiotic mouse model. Infect. Immun. 1979, 23, 403–411. [Google Scholar] [CrossRef]
- Yoo, J.Y.; Groer, M.; Dutra, S.V.O.; Sarkar, A.; McSkimming, D.I. Gut Microbiota and Immune System Interactions. Microorganisms 2020, 8, 1587. [Google Scholar] [CrossRef]
- Shah, T.; Shah, Z.; Baloch, Z.; Cui, X. The role of microbiota in respiratory health and diseases, particularly in tuberculosis. Biomed. Pharmacother. 2021, 143, 112108. [Google Scholar] [CrossRef]
- Dumas, A.; Corral, D.; Colom, A.; Levillain, F.; Peixoto, A.; Hudrisier, D.; Poquet, Y.; Neyrolles, O. The Host Microbiota Contributes to Early Protection Against Lung Colonization by Mycobacterium tuberculosis. Front. Immunol. 2018, 9, 2656. [Google Scholar] [CrossRef]
- Wozniak, H.; Beckmann, T.S.; Frohlich, L.; Soccorsi, T.; Le Terrier, C.; de Watteville, A.; Schrenzel, J.; Heidegger, C.P. The central and biodynamic role of gut microbiota in critically ill patients. Crit. Care 2022, 26, 250. [Google Scholar] [CrossRef]
- Abt, M.C.; Osborne, L.C.; Monticelli, L.A.; Doering, T.A.; Alenghat, T.; Sonnenberg, G.F.; Paley, M.A.; Antenus, M.; Williams, K.L.; Erikson, J.; et al. Commensal bacteria calibrate the activation threshold of innate antiviral immunity. Immunity 2012, 37, 158–170. [Google Scholar] [CrossRef]
- Bradley, K.C.; Finsterbusch, K.; Schnepf, D.; Crotta, S.; Llorian, M.; Davidson, S.; Fuchs, S.Y.; Staeheli, P.; Wack, A. Microbiota-Driven Tonic Interferon Signals in Lung Stromal Cells Protect from Influenza Virus Infection. Cell Rep. 2019, 28, 245–256.e244. [Google Scholar] [CrossRef]
- Brown, R.L.; Sequeira, R.P.; Clarke, T.B. The microbiota protects against respiratory infection via GM-CSF signaling. Nat. Commun. 2017, 8, 1512. [Google Scholar] [CrossRef]
- Yang, D.; Guo, X.; Huang, T.; Liu, C. The Role of Group 3 Innate Lymphoid Cells in Lung Infection and Immunity. Front. Cell. Infect. Microbiol. 2021, 11, 586471. [Google Scholar] [CrossRef]
- Vinolo, M.A.; Rodrigues, H.G.; Nachbar, R.T.; Curi, R. Regulation of inflammation by short chain fatty acids. Nutrients 2011, 3, 858–876. [Google Scholar] [CrossRef]
- Li, M.; van Esch, B.; Wagenaar, G.T.M.; Garssen, J.; Folkerts, G.; Henricks, P.A.J. Pro- and anti-inflammatory effects of short chain fatty acids on immune and endothelial cells. Eur. J. Pharmacol. 2018, 831, 52–59. [Google Scholar] [CrossRef] [PubMed]
- McAleer, J.P.; Kolls, J.K. Contributions of the intestinal microbiome in lung immunity. Eur. J. Immunol. 2018, 48, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Dickson, R.P.; Singer, B.H.; Newstead, M.W.; Falkowski, N.R.; Erb-Downward, J.R.; Standiford, T.J.; Huffnagle, G.B. Enrichment of the lung microbiome with gut bacteria in sepsis and the acute respiratory distress syndrome. Nat. Microbiol. 2016, 1, 16113. [Google Scholar] [CrossRef]
- Li, K.L.; Wang, B.Z.; Li, Z.P.; Li, Y.L.; Liang, J.J. Alterations of intestinal flora and the effects of probiotics in children with recurrent respiratory tract infection. World J. Pediatr. 2019, 15, 255–261. [Google Scholar] [CrossRef] [PubMed]
- Batra, P.; Soni, K.D.; Mathur, P. Efficacy of probiotics in the prevention of VAP in critically ill ICU patients: An updated systematic review and meta-analysis of randomized control trials. J. Intensive Care 2020, 8, 81. [Google Scholar] [CrossRef] [PubMed]
- Johnstone, J.; Meade, M.; Lauzier, F.; Marshall, J.; Duan, E.; Dionne, J.; Arabi, Y.M.; Heels-Ansdell, D.; Thabane, L.; Lamarche, D.; et al. Effect of Probiotics on Incident Ventilator-Associated Pneumonia in Critically Ill Patients: A Randomized Clinical Trial. JAMA 2021, 326, 1024–1033. [Google Scholar] [CrossRef] [PubMed]
- Hanada, S.; Pirzadeh, M.; Carver, K.Y.; Deng, J.C. Respiratory Viral Infection-Induced Microbiome Alterations and Secondary Bacterial Pneumonia. Front. Immunol. 2018, 9, 2640. [Google Scholar] [CrossRef]
- Charlson, E.S.; Bittinger, K.; Haas, A.R.; Fitzgerald, A.S.; Frank, I.; Yadav, A.; Bushman, F.D.; Collman, R.G. Topographical continuity of bacterial populations in the healthy human respiratory tract. Am. J. Respir. Crit. Care Med. 2011, 184, 957–963. [Google Scholar] [CrossRef]
- Bassis, C.M.; Tang, A.L.; Young, V.B.; Pynnonen, M.A. The nasal cavity microbiota of healthy adults. Microbiome 2014, 2, 27. [Google Scholar] [CrossRef]
- Bassis, C.M.; Erb-Downward, J.R.; Dickson, R.P.; Freeman, C.M.; Schmidt, T.M.; Young, V.B.; Beck, J.M.; Curtis, J.L.; Huffnagle, G.B. Analysis of the upper respiratory tract microbiotas as the source of the lung and gastric microbiotas in healthy individuals. mBio 2015, 6, e00037. [Google Scholar] [CrossRef]
- Man, W.H.; de Steenhuijsen Piters, W.A.; Bogaert, D. The microbiota of the respiratory tract: Gatekeeper to respiratory health. Nat. Rev. Microbiol. 2017, 15, 259–270. [Google Scholar] [CrossRef] [PubMed]
- Dickson, R.P.; Erb-Downward, J.R.; Freeman, C.M.; McCloskey, L.; Beck, J.M.; Huffnagle, G.B.; Curtis, J.L. Spatial Variation in the Healthy Human Lung Microbiome and the Adapted Island Model of Lung Biogeography. Ann. Am. Thorac. Soc. 2015, 12, 821–830. [Google Scholar] [CrossRef] [PubMed]
- Du, T.; Lei, A.; Zhang, N.; Zhu, C. The Beneficial Role of Probiotic Lactobacillus in Respiratory Diseases. Front. Immunol. 2022, 13, 908010. [Google Scholar] [CrossRef] [PubMed]
- Hori, T.; Kiyoshima, J.; Shida, K.; Yasui, H. Effect of intranasal administration of Lactobacillus casei Shirota on influenza virus infection of upper respiratory tract in mice. Clin. Diagn. Lab. Immunol. 2001, 8, 593–597. [Google Scholar] [CrossRef]
- Chong, H.X.; Yusoff, N.A.A.; Hor, Y.Y.; Lew, L.C.; Jaafar, M.H.; Choi, S.B.; Yusoff, M.S.B.; Wahid, N.; Abdullah, M.; Zakaria, N.; et al. Lactobacillus plantarum DR7 improved upper respiratory tract infections via enhancing immune and inflammatory parameters: A randomized, double-blind, placebo-controlled study. J. Dairy Sci. 2019, 102, 4783–4797. [Google Scholar] [CrossRef]
- Qi, X.; Qu, H.; Yang, D.; Zhou, L.; He, Y.W.; Yu, Y.; Qu, J.; Liu, J. Lower respiratory tract microbial composition was diversified in Pseudomonas aeruginosa ventilator-associated pneumonia patients. Respir. Res. 2018, 19, 139. [Google Scholar] [CrossRef]
- Zakharkina, T.; Martin-Loeches, I.; Matamoros, S.; Povoa, P.; Torres, A.; Kastelijn, J.B.; Hofstra, J.J.; de Wever, B.; de Jong, M.; Schultz, M.J.; et al. The dynamics of the pulmonary microbiome during mechanical ventilation in the intensive care unit and the association with occurrence of pneumonia. Thorax 2017, 72, 803–810. [Google Scholar] [CrossRef]
- Bai, C.; Liu, T.; Xu, J.; Ma, X.; Huang, L.; Liu, S.; Yu, H.; Chen, J.; Gu, X. Effect of High Calorie Diet on Intestinal Flora in LPS-Induced Pneumonia Rats. Sci. Rep. 2020, 10, 1701. [Google Scholar] [CrossRef]
- Sharma, G.; Garg, N.; Hasan, S.; Shirodkar, S. Prevotella: An insight into its characteristics and associated virulence factors. Microb. Pathog. 2022, 169, 105673. [Google Scholar] [CrossRef]
- Lin, W.; Zhang, Q.; Chen, Y.; Dong, B.; Xue, H.; Lei, H.; Lu, Y.; Wei, X.; Sun, P. Changes of the vaginal microbiota in HPV infection and cervical intraepithelial neoplasia: A cross-sectional analysis. Sci. Rep. 2022, 12, 2812. [Google Scholar] [CrossRef]
- Romani, L.; Del Chierico, F.; Macari, G.; Pane, S.; Ristori, M.V.; Guarrasi, V.; Gardini, S.; Pascucci, G.R.; Cotugno, N.; Perno, C.F.; et al. The Relationship Between Pediatric Gut Microbiota and SARS-CoV-2 Infection. Front. Cell. Infect. Microbiol. 2022, 12, 908492. [Google Scholar] [CrossRef] [PubMed]
- Winkler, E.S.; Shrihari, S.; Hykes, B.L., Jr.; Handley, S.A.; Andhey, P.S.; Huang, Y.S.; Swain, A.; Droit, L.; Chebrolu, K.K.; Mack, M.; et al. The Intestinal Microbiome Restricts Alphavirus Infection and Dissemination through a Bile Acid-Type I IFN Signaling Axis. Cell 2020, 182, 901–918.e918. [Google Scholar] [CrossRef] [PubMed]
- Hirayama, M.; Nishiwaki, H.; Hamaguchi, T.; Ito, M.; Ueyama, J.; Maeda, T.; Kashihara, K.; Tsuboi, Y.; Ohno, K. Intestinal Collinsella may mitigate infection and exacerbation of COVID-19 by producing ursodeoxycholate. PLoS ONE 2021, 16, e0260451. [Google Scholar] [CrossRef] [PubMed]
- Theriot, C.M.; Bowman, A.A.; Young, V.B. Antibiotic-Induced Alterations of the Gut Microbiota Alter Secondary Bile Acid Production and Allow for Clostridium difficile Spore Germination and Outgrowth in the Large Intestine. MSphere 2016, 1, 10–1128. [Google Scholar] [CrossRef]
- Allegretti, J.R.; Kearney, S.; Li, N.; Bogart, E.; Bullock, K.; Gerber, G.K.; Bry, L.; Clish, C.B.; Alm, E.; Korzenik, J.R. Recurrent Clostridium difficile infection associates with distinct bile acid and microbiome profiles. Aliment. Pharmacol. Ther. 2016, 43, 1142–1153. [Google Scholar] [CrossRef]
- Kim, M.; Qie, Y.; Park, J.; Kim, C.H. Gut Microbial Metabolites Fuel Host Antibody Responses. Cell Host Microbe 2016, 20, 202–214. [Google Scholar] [CrossRef]
- Rooks, M.G.; Garrett, W.S. Gut microbiota, metabolites and host immunity. Nat. Rev. Immunol. 2016, 16, 341–352. [Google Scholar] [CrossRef]
- Meganathan, R. Biosynthesis of menaquinone (vitamin K2) and ubiquinone (coenzyme Q): A perspective on enzymatic mechanisms. Vitam. Horm. 2001, 61, 173–218. [Google Scholar] [CrossRef]
- Popescu, A.; German, M. Vitamin K2 Holds Promise for Alzheimer’s Prevention and Treatment. Nutrients 2021, 13, 2206. [Google Scholar] [CrossRef]
- Li, Y.; Chen, J.P.; Duan, L.; Li, S. Effect of vitamin K2 on type 2 diabetes mellitus: A review. Diabetes Res. Clin. Pract. 2018, 136, 39–51. [Google Scholar] [CrossRef]
- Bai, X.; Narayanan, A.; Skagerberg, M.; Cena-Diez, R.; Giske, C.G.; Stralin, K.; Sonnerborg, A. Characterization of the Upper Respiratory Bacterial Microbiome in Critically Ill COVID-19 Patients. Biomedicines 2022, 10, 982. [Google Scholar] [CrossRef] [PubMed]
- Karl, J.P.; Meydani, M.; Barnett, J.B.; Vanegas, S.M.; Barger, K.; Fu, X.; Goldin, B.; Kane, A.; Rasmussen, H.; Vangay, P.; et al. Fecal concentrations of bacterially derived vitamin K forms are associated with gut microbiota composition but not plasma or fecal cytokine concentrations in healthy adults. Am. J. Clin. Nutr. 2017, 106, 1052–1061. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, L.; Wei, C.; Wang, X.; Li, R.; Xu, X.; Zhang, Y.; Geng, G.; Dang, K.; Ming, Z.; et al. Vitamin K2 supplementation improves impaired glycemic homeostasis and insulin sensitivity for type 2 diabetes through gut microbiome and fecal metabolites. BMC Med. 2023, 21, 174. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Infections | Exposures | nSNP | MR Analysis | Cochran’s Q Test | MR Egger Intercept | Causal Direction * | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Method | b | SE | p Value | OR | 95%CI | Value | p Value | Value | p Value | ||||
| URTIs | Family Lactobacillaceae | 6 | IVW | −0.118 | 0.039 | 0.002 | 0.889 | 0.824–0.959 | 2.958 | 0.706 | −2.56 × 10−3 | 0.907 | TRUE |
| MR Egger | −0.103 | 0.129 | 0.470 | 0.903 | 0.701–1.162 | 2.942 | 0.568 | ||||||
| Weighted median | −0.128 | 0.053 | 0.016 | 0.880 | 0.793–0.976 | ||||||||
| Simple mode | −0.141 | 0.076 | 0.122 | 0.868 | 0.748–1.007 | ||||||||
| Weighted mode | −0.138 | 0.066 | 0.092 | 0.871 | 0.765–0.992 | ||||||||
| Genus Family XIII AD3011 | 12 | IVW | −0.136 | 0.046 | 0.003 | 0.873 | 0.798–0.955 | 9.950 | 0.535 | −2.18 × 10−2 | 0.228 | TRUE | |
| MR Egger | 0.125 | 0.208 | 0.562 | 1.133 | 0.754–1.704 | 8.297 | 0.600 | ||||||
| Weighted median | −0.076 | 0.064 | 0.234 | 0.927 | 0.818–1.05 | ||||||||
| Simple mode | −0.045 | 0.097 | 0.651 | 0.956 | 0.791–1.156 | ||||||||
| Weighted mode | −0.049 | 0.085 | 0.576 | 0.952 | 0.807–1.124 | ||||||||
| LRTIs | Order Bacillales | 7 | IVW | 0.001 | 0.001 | 0.020 | 1.001 | 1.000–1.002 | 5.924 | 0.432 | −9.13 × 10−5 | 0.897 | TRUE |
| MR Egger | 0.002 | 0.005 | 0.703 | 1.002 | 0.992–1.012 | 5.902 | 0.316 | ||||||
| Weighted median | 0.001 | 0.001 | 0.155 | 1.001 | 1.000–1.003 | ||||||||
| Simple mode | 0.001 | 0.001 | 0.334 | 1.001 | 0.999–1.004 | ||||||||
| Weighted mode | 0.001 | 0.001 | 0.307 | 1.001 | 0.999–1.003 | ||||||||
| Genus Ruminococcaceae UCG009 | 8 | IVW | −0.003 | 0.001 | 0.002 | 0.997 | 0.996–0.999 | 2.343 | 0.938 | 2.96 × 10−4 | 0.739 | TRUE | |
| MR Egger | −0.006 | 0.010 | 0.572 | 0.994 | 0.975–1.014 | 2.222 | 0.898 | ||||||
| Weighted median | −0.002 | 0.001 | 0.037 | 0.998 | 0.996–1.000 | ||||||||
| Simple mode | −0.002 | 0.002 | 0.223 | 0.998 | 0.995–1.001 | ||||||||
| Weighted mode | −0.002 | 0.001 | 0.188 | 0.998 | 0.995–1.001 | ||||||||
| Genus Paraprevotella | 7 | IVW | 0.003 | 0.001 | 0.001 | 1.003 | 1.001–1.004 | 2.593 | 0.858 | −1.00 × 10−4 | 0.844 | TRUE | |
| MR Egger | 0.004 | 0.005 | 0.495 | 1.004 | 0.994–1.014 | 2.55 | 0.769 | ||||||
| Weighted median | 0.002 | 0.001 | 0.034 | 1.002 | 1.000–1.005 | ||||||||
| Simple mode | 0.003 | 0.002 | 0.176 | 1.003 | 0.999–1.006 | ||||||||
| Weighted mode | 0.003 | 0.002 | 0.156 | 1.003 | 0.999–1.006 | ||||||||
| Genus Alistipes | 7 | IVW | −0.004 | 0.001 | 0.001 | 0.996 | 0.993–0.998 | 3.617 | 0.728 | 1.69 × 10−4 | 0.750 | TRUE | |
| MR Egger | −0.007 | 0.009 | 0.443 | 0.993 | 0.975–1.01 | 3.503 | 0.623 | ||||||
| Weighted median | −0.005 | 0.002 | 0.010 | 0.995 | 0.992–0.999 | ||||||||
| Simple mode | −0.006 | 0.003 | 0.063 | 0.994 | 0.989–0.999 | ||||||||
| Weighted mode | −0.002 | 0.002 | 0.361 | 0.998 | 0.993–1.002 | ||||||||
| LRTIs | Deoxycholate | 11 | IVW | −0.004 | 0.002 | 0.005 | 0.996 | 0.993–0.999 | 11.086 | 0.351 | 3.31 × 10−4 | 0.149 | TRUE |
| MR Egger | −0.012 | 0.005 | 0.042 | 0.988 | 0.978–0.998 | 8.596 | 0.475 | ||||||
| Weighted median | −0.003 | 0.002 | 0.214 | 0.997 | 0.993–1.002 | ||||||||
| Simple mode | 0.000 | 0.003 | 0.909 | 1.000 | 0.994–1.007 | ||||||||
| Weighted mode | −0.001 | 0.004 | 0.767 | 0.999 | 0.992–1.006 | ||||||||
| LRTIs | Menaquinol 8 biosynthesis II | 3 | IVW | 0.002 | 0.001 | 0.002 | 1.002 | 1.001–1.003 | 0.187 | 0.911 | −1.83 × 10−5 | 0.980 | TRUE |
| MR Egger | 0.002 | 0.003 | 0.645 | 1.002 | 0.996–1.008 | 0.186 | 0.667 | ||||||
| Weighted median | 0.002 | 0.001 | 0.017 | 1.002 | 1–1.003 | ||||||||
| Simple mode | 0.002 | 0.001 | 0.159 | 1.002 | 1–1.004 | ||||||||
| Weighted mode | 0.002 | 0.001 | 0.160 | 1.002 | 1–1.004 | ||||||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, S.; Li, J.; Zhu, Z.; Liu, X.; Shen, T.; Wang, Y.; Ma, Q.; Wang, X.; Yang, G.; Guo, G.; et al. Gut Microbiota and Respiratory Infections: Insights from Mendelian Randomization. Microorganisms 2023, 11, 2108. https://doi.org/10.3390/microorganisms11082108
Huang S, Li J, Zhu Z, Liu X, Shen T, Wang Y, Ma Q, Wang X, Yang G, Guo G, et al. Gut Microbiota and Respiratory Infections: Insights from Mendelian Randomization. Microorganisms. 2023; 11(8):2108. https://doi.org/10.3390/microorganisms11082108
Chicago/Turabian StyleHuang, Shengyu, Jiaqi Li, Zhihao Zhu, Xiaobin Liu, Tuo Shen, Yusong Wang, Qimin Ma, Xin Wang, Guangping Yang, Guanghua Guo, and et al. 2023. "Gut Microbiota and Respiratory Infections: Insights from Mendelian Randomization" Microorganisms 11, no. 8: 2108. https://doi.org/10.3390/microorganisms11082108