
Revisiting Microbial Diversity in Hypersaline Microbial Mats from Guerrero Negro for a Better Understanding of Methanogenic Archaeal Communities

 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Fieldwork
2.2. Library Preparation of 16S rRNA and mcrA Genes
2.3. High-Throughput Sequencing
2.4. Bioinformatic Analyses of 16S rRNA Metaprofiling
2.5. Processing the mcrA Amplicon Metaprofiling
2.6. Whole Metagenome Shotgun Data
3. Results
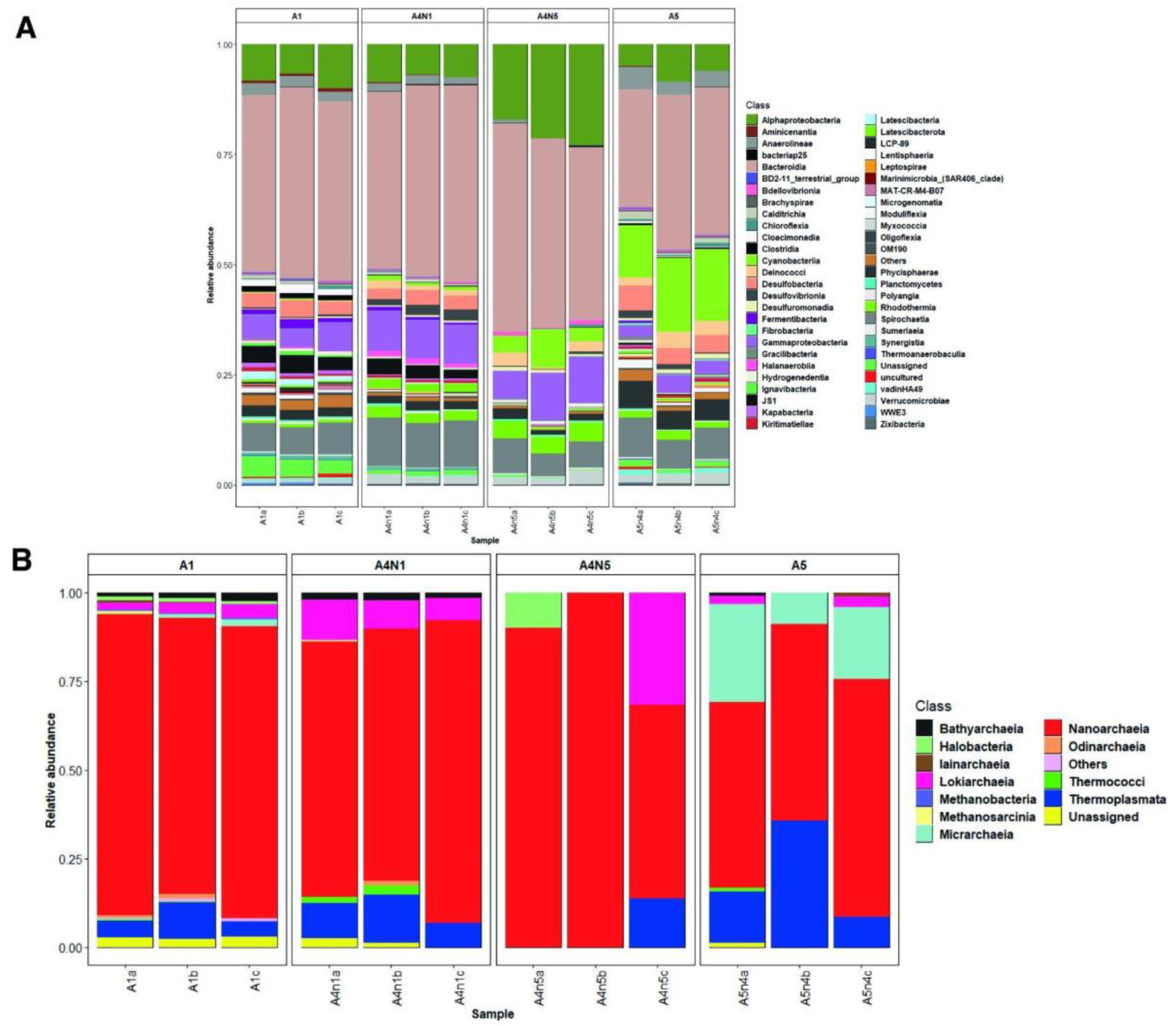
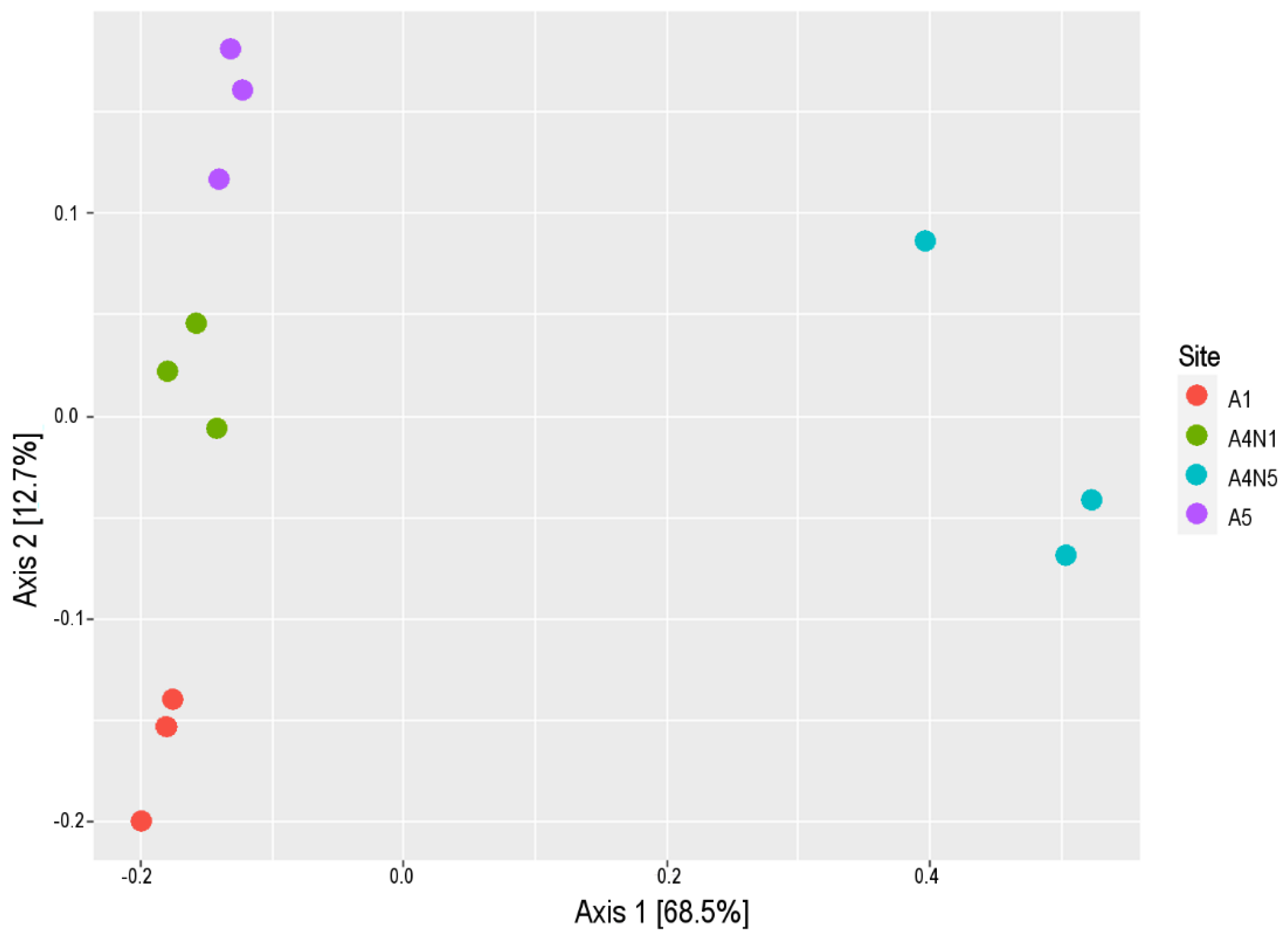
3.1. Environmental Variables and Microbial Diversity
3.2. Composition and Phylogeny of Methanogenic Archaea
3.3. Insights into the Metabolic Pathway of Methanogens
4. Discussion
4.1. Methanogenic Diversity in Hypersaline Microbial Mats
4.2. Methanogenic Metabolism in Hypersaline Microbial Mats
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Huang, W.-C.; Liu, Y.; Zhang, X.; Zhang, C.-J.; Zou, D.; Zheng, S.; Xu, W.; Luo, Z.; Liu, F.; Li, M. Comparative genomic analysis reveals metabolic flexibility of Woesearchaeota. Nat. Commun. 2021, 12, 5281. [Google Scholar] [CrossRef]
- Adam, P.S.; Borrel, G.; Brochier-Armanet, C.; Gribaldo, S. The growing tree of Archaea: New perspectives on their diversity, evolution and ecology. ISME J. 2017, 11, 2407–2425. [Google Scholar] [CrossRef] [Green Version]
- Spang, A.; Caceres, E.F.; Ettema, T.J.G. Genomic exploration of the diversity, ecology, and evolution of the archaeal domain of life. Science 2017, 357, eaaf3883. [Google Scholar] [CrossRef] [Green Version]
- Baker, B.J.; De Anda, V.; Seitz, K.W.; Dombrowski, N.; Santoro, A.E.; Lloyd, K.G. Diversity, ecology and evolution of Archaea. Nat. Microbiol. 2020, 5, 887–900. [Google Scholar] [CrossRef] [PubMed]
- Orphan, V.J.; Jahnke, L.L.; Embaye, T.; Turk, K.A.; Pernthaler, A.; Summons, R.E.; Marais, D.J.D. Characterization and spatial distribution of methanogens and methanogenic biosignatures in hypersaline microbial mats of Baja California. Geobiology 2008, 6, 376–393. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.M.; Green, S.J.; Kelley, C.A.; Prufert-Bebout, L.; Bebout, B.M. Shifts in methanogen community structure and function associated with long-term manipulation of sulfate and salinity in a hypersaline microbial mat. Environ. Microbiol. 2007, 10, 386–394. [Google Scholar] [CrossRef] [PubMed]
- Robertson, C.E.; Spear, J.R.; Harris, J.K.; Pace, N.R. Diversity and Stratification of Archaea in a Hypersaline Microbial Mat. Appl. Environ. Microbiol. 2009, 75, 1801–1810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García-Maldonado, J.Q.; Bebout, B.M.; Everroad, R.C.; López-Cortés, A. Evidence of Novel Phylogenetic Lineages of Methanogenic Archaea from Hypersaline Microbial Mats. Microb. Ecol. 2014, 69, 106–117. [Google Scholar] [CrossRef] [PubMed]
- Wong, H.L.; Visscher, P.T.; Iii, R.A.W.; Smith, D.-L.; Patterson, M.M.; Burns, B.P. Dynamics of archaea at fine spatial scales in Shark Bay mat microbiomes. Sci. Rep. 2017, 7, srep46160. [Google Scholar] [CrossRef] [Green Version]
- García-Maldonado, J.Q.; Escobar-Zepeda, A.; Raggi, L.; Bebout, B.M.; Sanchez-Flores, A.; López-Cortés, A. Bacterial and archaeal profiling of hypersaline microbial mats and endoevaporites, under natural conditions and methanogenic microcosm experiments. Extremophiles 2018, 22, 903–916. [Google Scholar] [CrossRef]
- Paul, K.; Nonoh, J.O.; Mikulski, L.; Brune, A. “Methanoplasmatales,” Thermoplasmatales-Related Archaea in Termite Guts and Other Environments, Are the Seventh Order of Methanogens. Appl. Environ. Microbiol. 2012, 78, 8245–8253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borrel, G.; McCann, A.; Deane, J.; Neto, M.C.; Lynch, D.B.; Brugère, J.-F.; O’Toole, P.W. Genomics and metagenomics of trimethylamine-utilizing Archaea in the human gut microbiome. ISME J. 2017, 11, 2059–2074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Söllinger, A.; Schwab, C.; Weinmaier, T.; Loy, A.; Tveit, A.T.; Schleper, C.; Urich, T. Phylogenetic and genomic analysis of Methanomassiliicoccales in wetlands and animal intestinal tracts reveals clade-specific habitat preferences. FEMS Microbiol. Ecol. 2015, 92, fiv149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berghuis, B.A.; Yu, F.B.; Schulz, F.; Blainey, P.C.; Woyke, T.; Quake, S.R. Hydrogenotrophic methanogenesis in archaeal phylum Verstraetearchaeota reveals the shared ancestry of all methanogens. Proc. Natl. Acad. Sci. USA 2019, 116, 5037–5044. [Google Scholar] [CrossRef] [Green Version]
- Söllinger, A.; Urich, T. Methylotrophic methanogens everywhere—Physiology and ecology of novel players in global methane cycling. Biochem. Soc. Trans. 2019, 47, 1895–1907. [Google Scholar] [CrossRef]
- Kallistova, A.; Merkel, A.; Kanapatskiy, T.; Boltyanskaya, Y.; Tarnovetskii, I.; Perevalova, A.; Kevbrin, V.; Samylina, O.; Pimenov, N. Methanogenesis in the Lake Elton saline aquatic system. Extremophiles 2020, 24, 657–672. [Google Scholar] [CrossRef]
- Lang, K.; Schuldes, J.; Klingl, A.; Poehlein, A.; Daniel, R.; Brune, A. New Mode of Energy Metabolism in the Seventh Order of Methanogens as Revealed by Comparative Genome Analysis of “Candidatus Methanoplasma termitum”. Appl. Environ. Microbiol. 2015, 81, 1338–1352. [Google Scholar] [CrossRef] [Green Version]
- Sorokin, D.Y.; Makarova, K.S.; Abbas, B.; Ferrer, M.; Golyshin, P.N.; Galinski, E.A.; Ciordia, S.; Mena, M.C.; Merkel, A.Y.; Wolf, Y.I.; et al. Discovery of extremely halophilic, methyl-reducing euryarchaea provides insights into the evolutionary origin of methanogenesis. Nat. Microbiol. 2017, 2, 17081. [Google Scholar] [CrossRef] [Green Version]
- Evans, P.; Parks, D.H.; Chadwick, G.; Robbins, S.J.; Orphan, V.; Golding, S.D.; Tyson, G.W. Methane metabolism in the archaeal phylum Bathyarchaeota revealed by genome-centric metagenomics. Science 2015, 350, 434–438. [Google Scholar] [CrossRef] [Green Version]
- Vanwonterghem, I.; Evans, P.N.; Parks, D.H.; Jensen, P.D.; Woodcroft, B.J.; Hugenholtz, P.; Tyson, G.W. Methylotrophic methanogenesis discovered in the archaeal phylum Verstraetearchaeota. Nat. Microbiol. 2016, 1, 16170. [Google Scholar] [CrossRef] [Green Version]
- Sprenger, W.W.; Van Belzen, M.C.; Rosenberg, J.; Hackstein, J.H.; Keltjens, J.T. Methanomicrococcus blatticola gen. nov., sp. nov., a methanol- and methylamine-reducing methanogen from the hindgut of the cockroach Periplaneta americana. Int. J. Syst. Evol. Microbiol. 2000, 50, 1989–1999. [Google Scholar] [CrossRef] [Green Version]
- Parada, A.E.; Needham, D.M.; Fuhrman, J.A. Every base matters: Assessing small subunit rRNA primers for marine microbiomes with mock communities, time series and global field samples. Environ. Microbiol. 2016, 18, 1403–1414. [Google Scholar] [CrossRef]
- Angel, R.; Claus, P.; Conrad, R. Methanogenic archaea are globally ubiquitous in aerated soils and become active under wet anoxic conditions. ISME J. 2011, 6, 847–862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Gonzalez Peña, A.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Callahan, B.J.; Mcmurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Callahan, B.J.; McMurdie, P.J.; Holmes, S.P. Exact sequence variants should replace operational taxonomic units in marker-gene data analysis. ISME J. 2017, 11, 2639–2643. [Google Scholar] [CrossRef] [Green Version]
- Rognes, T.; Flouri, T.; Nichols, B.; Quince, C.; Mahé, F. VSEARCH: A versatile open source tool for metagenomics. PeerJ 2016, 4, e2584. [Google Scholar] [CrossRef] [Green Version]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2—Approximately Maximum-Likelihood Trees for Large Alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef]
- McMurdie, P.J.; Holmes, S. phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef] [Green Version]
- Oksanen, J.; Kindt, R.; Legendre, P.; O’Hara, B.; Simpson, G.L.; Solymos, P.; Stevens, M.H.H.; Wagn, H. The vegan package. Community Ecol. Package 2011, 10, 719. [Google Scholar]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: Berlin/Heidelberg, Germany, 2009; p. 213. [Google Scholar] [CrossRef]
- Lozupone, C.; Knight, R. UniFrac: A New Phylogenetic Method for Comparing Microbial Communities. Appl. Environ. Microbiol. 2005, 71, 8228–8235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, M.J. A new method for non-parametric multivariate analysis of variance. Austral Ecol. 2001, 26, 32–46. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Magoč, T.; Salzberg, S.L. FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef] [Green Version]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [Green Version]
- Cline, K. Taxtastic: Create and Maintain Phylogenetic “Reference Packages” of Biological Sequences. n.d. Github. 2019. Available online: https://github.com/fhcrc/taxtastic (accessed on 24 August 2021).
- Matsen, F.A.; Kodner, R.B.; Armbrust, E.V. pplacer: Linear time maximum-likelihood and Bayesian phylogenetic placement of sequences onto a fixed reference tree. BMC Bioinform. 2010, 11, 538. [Google Scholar] [CrossRef] [Green Version]
- Waterhouse, A.M.; Procter, J.B.; Martin, D.M.A.; Clamp, M.; Barton, G.J. Jalview Version 2—A multiple sequence alignment editor and analysis workbench. Bioinformatics 2009, 25, 1189–1191. [Google Scholar] [CrossRef] [Green Version]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef]
- Shen, W.; Ren, H. TaxonKit: A practical and efficient NCBI taxonomy toolkit. J. Genet. Genom. 2021, 48, 844–850. [Google Scholar] [CrossRef] [PubMed]
- Maza-Márquez, P.; Lee, M.D.; Detweiler, A.M.; Bebout, B.M. Millimeter-scale vertical partitioning of nitrogen cycling in hypersaline mats reveals prominence of genes encoding multi-heme and prismane proteins. ISME J. 2021, 16, 1119–1129. [Google Scholar] [CrossRef] [PubMed]
- Shaffer, M.; Borton, M.A.; McGivern, B.B.; Zayed, A.A.; La Rosa, S.L.; Solden, L.M.; Liu, P.; Narrowe, A.B.; Rodríguez-Ramos, J.; Bolduc, B.; et al. DRAM for distilling microbial metabolism to automate the curation of microbiome function. Nucleic Acids Res. 2020, 48, 8883–8900. [Google Scholar] [CrossRef] [PubMed]
- Cantalapiedra, C.P.; Hernández-Plaza, A.; Letunic, I.; Bork, P.; Huerta-Cepas, J. eggNOG-mapper v2: Functional Annotation, Orthology Assignments, and Domain Prediction at the Metagenomic Scale. Mol. Biol. Evol. 2021, 38, 5825–5829. [Google Scholar] [CrossRef] [PubMed]
- Techa-Angkoon, P.; Sun, Y.; Lei, J. A sensitive short read homology search tool for paired-end read sequencing data. BMC Bioinform. 2017, 18, 414. [Google Scholar] [CrossRef] [Green Version]
- Shu, W.-S.; Huang, L.-N. Microbial diversity in extreme environments. Nat. Rev. Genet. 2021, 20, 219–235. [Google Scholar] [CrossRef]
- Marais, D.J.D. Microbial mats and the early evolution of life. Trends Ecol. Evol. 1990, 5, 140–144. [Google Scholar] [CrossRef]
- Ley, R.E.; Harris, J.K.; Wilcox, J.; Spear, J.R.; Miller, S.R.; Bebout, B.M.; Maresca, J.A.; Bryant, D.A.; Sogin, M.L.; Pace, N.R. Unexpected Diversity and Complexity of the Guerrero Negro Hypersaline Microbial Mat. Appl. Environ. Microbiol. 2006, 72, 3685–3695. [Google Scholar] [CrossRef] [Green Version]
- Harris, J.K.; Caporaso, J.G.; Walker, J.J.; Spear, J.R.; Gold, N.J.; Robertson, C.E.; Hugenholtz, P.; Goodrich, J.; McDonald, D.; Knights, D.; et al. Phylogenetic stratigraphy in the Guerrero Negro hypersaline microbial mat. ISME J. 2012, 7, 50–60. [Google Scholar] [CrossRef] [Green Version]
- Baker, B.J.; Comolli, L.R.; Dick, G.J.; Hauser, L.J.; Hyatt, D.; Dill, B.D.; Land, M.L.; VerBerkmoes, N.C.; Hettich, R.L.; Banfield, J.F. Enigmatic, ultrasmall, uncultivated Archaea. Proc. Natl. Acad. Sci. USA 2010, 107, 8806–8811. [Google Scholar] [CrossRef] [Green Version]
- Castelle, C.J.; Wrighton, K.C.; Thomas, B.C.; Hug, L.A.; Brown, C.T.; Wilkins, M.J.; Frischkorn, K.R.; Tringe, S.G.; Singh, A.; Markillie, L.M.; et al. Genomic Expansion of Domain Archaea Highlights Roles for Organisms from New Phyla in Anaerobic Carbon Cycling. Curr. Biol. 2015, 25, 690–701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wurch, L.; Giannone, R.J.; Belisle, B.S.; Swift, C.; Utturkar, S.; Hettich, R.L.; Reysenbach, A.-L.; Podar, M. Genomics-informed isolation and characterization of a symbiotic Nanoarchaeota system from a terrestrial geothermal environment. Nat. Commun. 2016, 7, 12115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Golyshina, O.V.; Bargiela, R.; Toshchakov, S.V.; Chernyh, N.A.; Ramayah, S.; Korzhenkov, A.A.; Kublanov, I.V.; Golyshin, P.N. Diversity of “Ca. Micrarchaeota” in Two Distinct Types of Acidic Environments and Their Associations with Thermoplasmatales. Genes 2019, 10, 461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Comolli, L.R.; Banfield, J.F. Inter-species interconnections in acid mine drainage microbial communities. Front. Microbiol. 2014, 5, 367. [Google Scholar] [CrossRef] [Green Version]
- Kadnikov, V.V.; Savvichev, A.S.; Mardanov, A.V.; Beletsky, A.V.; Chupakov, A.V.; Kokryatskaya, N.M.; Pimenov, N.V.; Ravin, N.V. Metabolic Diversity and Evolutionary History of the Archaeal Phylum “ Candidatus Micrarchaeota” Uncovered from a Freshwater Lake Metagenome. Appl. Environ. Microbiol. 2020, 86, e02199-20. [Google Scholar] [CrossRef]
- Chen, L.-X.; Méndez-García, C.; Dombrowski, N.; Servín-Garcidueñas, L.E.; Eloe-Fadrosh, E.A.; Fang, B.-Z.; Luo, Z.-H.; Tan, S.; Zhi, X.-Y.; Hua, Z.-S.; et al. Metabolic versatility of small archaea Micrarchaeota and Parvarchaeota. ISME J. 2017, 12, 756–775. [Google Scholar] [CrossRef]
- Liu, X.; Li, M.; Castelle, C.; Probst, A.; Zhou, Z.; Pan, J.; Liu, Y.; Banfield, J.; Gu, J.-D. Insights into the ecology, evolution, and metabolism of the widespread Woesearchaeotal lineages. Microbiome 2018, 6, 102. [Google Scholar] [CrossRef]
- Spang, A.; Saw, J.H.; Jørgensen, S.L.; Zaremba-Niedzwiedzka, K.; Martijn, J.; Lind, A.E.; van Eijk, R.; Schleper, C.; Guy, L.; Ettema, T.J.G. Complex archaea that bridge the gap between prokaryotes and eukaryotes. Nature 2015, 521, 173–179. [Google Scholar] [CrossRef] [Green Version]
- Zaremba-Niedzwiedzka, K.; Caceres, E.F.; Saw, J.H.; Bäckström, D.; Juzokaite, L.; Vancaester, E.; Seitz, K.W.; Anantharaman, K.; Starnawski, P.; Kjeldsen, K.U.; et al. Asgard archaea illuminate the origin of eukaryotic cellular complexity. Nature 2017, 541, 353–358. [Google Scholar] [CrossRef] [Green Version]
- Lloyd, K. Beyond known methanogens. Science 2015, 350, 384. [Google Scholar] [CrossRef]
- Lynch, M.D.J.; Neufeld, J.D. Ecology and exploration of the rare biosphere. Nat. Rev. Genet. 2015, 13, 217–229. [Google Scholar] [CrossRef] [PubMed]
- Medina-Chávez, N.O.; Travisano, M. Archaeal Communities: The Microbial Phylogenomic Frontier. Front. Genet. 2022, 12, 693193. [Google Scholar] [CrossRef] [PubMed]
- García-Maldonado, J.Q.; Bebout, B. Phylogenetic diversity of methyl-coenzyme M reductase (mcrA) gene and methanogenesis from trimethylamine in hypersaline environments. Int. Microbiol. 2012, 15, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Wilkins, D.; Lu, X.-Y.; Shen, Z.; Chen, J.; Lee, P.K.H. Pyrosequencing of mcrA and Archaeal 16S rRNA Genes Reveals Diversity and Substrate Preferences of Methanogen Communities in Anaerobic Digesters. Appl. Environ. Microbiol. 2015, 81, 604–613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katayama, T.; Yoshioka, H.; Kaneko, M.; Amo, M.; Fujii, T.; Takahashi, H.A.; Yoshida, S.; Sakata, S. Cultivation and biogeochemical analyses reveal insights into methanogenesis in deep subseafloor sediment at a biogenic gas hydrate site. ISME J. 2022, 16, 1464–1472. [Google Scholar] [CrossRef] [PubMed]
- Sorokin, D.Y.; Merkel, A.Y.; Abbas, B.; Makarova, K.S.; Rijpstra, W.I.C.; Koenen, M.; Damsté, J.S.S.; Galinski, E.A.; Koonin, E.V.; Van Loosdrecht, M.C.M. Methanonatronarchaeum thermophilum gen. nov., sp. nov. and ‘Candidatus Methanohalarchaeum thermophilum’, extremely halo(natrono)philic methyl-reducing methanogens from hypersaline lakes comprising a new euryarchaeal class Methanonatronarchaeia classis nov. Int. J. Syst. Evol. Microbiol. 2018, 68, 2199–2208. [Google Scholar] [CrossRef] [PubMed]
- Beaudoin, C.S. Use of Stable Carbon Isotopes to Assess Anaerobic and Aerobic Methane Oxidation in Hypersaline Ponds. Master’s Thesis, University of Missouri, Columbia, MO, USA, 2015. [Google Scholar]
- Camp, H.J.M.O.D.; Islam, T.; Stott, M.B.; Harhangi, H.R.; Hynes, A.; Schouten, S.; Jetten, M.S.M.; Birkeland, N.-K.; Pol, A.; Dunfield, P.F. Environmental, genomic and taxonomic perspectives on methanotrophic Verrucomicrobia. Environ. Microbiol. Rep. 2009, 1, 293–306. [Google Scholar] [CrossRef]
- Bay, S.K.; Dong, X.; Bradley, J.A.; Leung, P.M.; Grinter, R.; Jirapanjawat, T.; Arndt, S.K.; Cook, P.L.M.; LaRowe, D.E.; Nauer, P.A.; et al. Trace gas oxidizers are widespread and active members of soil microbial communities. Nat. Microbiol. 2021, 6, 246–256. [Google Scholar] [CrossRef]
- Kelley, C.A.; Poole, J.A.; Tazaz, A.M.; Chanton, J.P.; Bebout, B.M. Substrate Limitation for Methanogenesis in Hypersaline Environments. Astrobiology 2012, 12, 89–97. [Google Scholar] [CrossRef]
- Kelley, C.A.; Chanton, J.P.; Bebout, B.M. Rates and pathways of methanogenesis in hypersaline environments as determined by 13C-labeling. Biogeochemistry 2015, 126, 329–341. [Google Scholar] [CrossRef]
- Borrel, G.; Harris, H.M.B.; Tottey, W.; Mihajlovski, A.; Parisot, N.; Peyretaillade, E.; Peyret, P.; Gribaldo, S.; O’Toole, P.; Brugère, J.-F. Genome Sequence of “Candidatus Methanomethylophilus alvus” Mx1201, a Methanogenic Archaeon from the Human Gut Belonging to a Seventh Order of Methanogens. J. Bacteriol. 2012, 194, 6944–6945. [Google Scholar] [CrossRef] [Green Version]
- Dridi, B.; Fardeau, M.-L.; Ollivier, B.; Raoult, D.; Drancourt, M. Methanomassiliicoccus luminyensis gen. nov., sp. nov., a methanogenic archaeon isolated from human faeces. Int. J. Syst. Evol. Microbiol. 2012, 62, 1902–1907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borrel, G.; Harris, H.M.B.; Parisot, N.; Gaci, N.; Tottey, W.; Mihajlovski, A.; Deane, J.; Gribaldo, S.; Bardot, O.; Peyretaillade, E.; et al. Genome Sequence of “ Candidatus Methanomassiliicoccus intestinalis” Issoire-Mx1, a Third Thermoplasmatales-Related Methanogenic Archaeon from Human Feces. Genome Announc. 2013, 1, e00453-13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, M.; Yin, X.; Tang, X.; Zhang, C.; Zheng, Q.; Li, M. Metatranscriptomics reveals different features of methanogenic archaea among global vegetated coastal ecosystems. Sci. Total. Environ. 2021, 802, 149848. [Google Scholar] [CrossRef] [PubMed]
- Hua, Z.-S.; Wang, Y.-L.; Evans, P.N.; Qu, Y.-N.; Goh, K.M.; Rao, Y.-Z.; Qi, Y.-L.; Li, Y.-X.; Huang, M.-J.; Jiao, J.-Y.; et al. Insights into the ecological roles and evolution of methyl-coenzyme M reductase-containing hot spring Archaea. Nat. Commun. 2019, 10, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parks, D.H.; Rinke, C.; Chuvochina, M.; Chaumeil, P.-A.; Woodcroft, B.J.; Evans, P.N.; Hugenholtz, P.; Tyson, G.W. Recovery of nearly 8,000 metagenome-assembled genomes substantially expands the tree of life. Nat. Microbiol. 2017, 2, 1533–1542. [Google Scholar] [CrossRef]
- Zhang, C.-J.; Pan, J.; Liu, Y.; Duan, C.-H.; Li, M. Genomic and transcriptomic insights into methanogenesis potential of novel methanogens from mangrove sediments. Microbiome 2020, 8, 94. [Google Scholar] [CrossRef]
- Thomas, F.; Hehemann, J.-H.; Rebuffet, E.; Czjzek, M.; Michel, G. Environmental and Gut Bacteroidetes: The Food Connection. Front. Microbiol. 2011, 2, 93. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Garcia, M.; Brazel, D.; Swan, B.; Arnosti, C.; Chain, P.; Reitenga, K.G.; Xie, G.; Poulton, N.; Gomez, M.L.; Masland, D.E.D.; et al. Capturing Single Cell Genomes of Active Polysaccharide Degraders: An Unexpected Contribution of Verrucomicrobia. PLoS ONE 2012, 7, e35314. [Google Scholar] [CrossRef] [Green Version]
- Burnet, M.C.; Dohnalkova, A.C.; Neumann, A.P.; Lipton, M.S.; Smith, R.D.; Suen, G.; Callister, S.J. Evaluating Models of Cellulose Degradation by Fibrobacter succinogenes S85. PLoS ONE 2015, 10, e0143809. [Google Scholar] [CrossRef]
- Meegoda, J.N.; Li, B.; Patel, K.; Wang, L.B. A Review of the Processes, Parameters, and Optimization of Anaerobic Digestion. Int. J. Environ. Res. Public Health 2018, 15, 2224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Javed, S.; Azeem, F.; Hussain, S.; Rasul, I.; Siddique, M.H.; Riaz, M.; Afzal, M.; Kouser, A.; Nadeem, H. Bacterial lipases: A review on purification and characterization. Prog. Biophys. Mol. Biol. 2017, 132, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Kirkegaard, R.H.; Dueholm, M.S.; McIlroy, S.J.; Nierychlo, M.; Karst, S.M.; Albertsen, M.; Nielsen, P.H. Genomic insights into members of the candidate phylum Hyd24-12 common in mesophilic anaerobic digesters. ISME J. 2016, 10, 2352–2364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waite, D.W.; Chuvochina, M.; Pelikan, C.; Parks, D.H.; Yilmaz, P.; Wagner, M.; Loy, A.; Naganuma, T.; Nakai, R.; Whitman, W.B.; et al. Proposal to reclassify the proteobacterial classes Deltaproteobacteria and Oligoflexia, and the phylum Thermodesulfobacteria into four phyla reflecting major functional capabilities. Int. J. Syst. Evol. Microbiol. 2020, 70, 5972–6016. [Google Scholar] [CrossRef]
- Oren, A. Formation and breakdown of glycine betaine and trimethylamine in hypersaline environments. Antonie Leeuwenhoek 1990, 58, 291–298. [Google Scholar] [CrossRef]
- Tsai, C.-R.; Garcia, J.-L.; Patel, B.K.C.; Cayol, J.-L.; Baresi, L.; Mah, R.A. Haloanaerobium alcaliphilum sp. nov., an Anaerobic Moderate Halophile from the Sediments of Great Salt Lake, Utah. Int. J. Syst. Evol. Microbiol. 1995, 45, 301–307. [Google Scholar] [CrossRef] [Green Version]
- King, G.M. Utilization of hydrogen, acetate, and “noncompetitive”; substrates by methanogenic bacteria in marine sediments. Geomicrobiol. J. 1984, 3, 275–306. [Google Scholar] [CrossRef]
- Barrett, E.L.; Kwan, H.S. Bacterial reduction of trimethylamine oxide. Annu. Rev. Microbiol. 1985, 39, 131–149. [Google Scholar] [CrossRef]
- Kröninger, L.; Gottschling, J.; Deppenmeier, U. Growth Characteristics of Methanomassiliicoccus luminyensis and Expression of Methyltransferase Encoding Genes. Archaea 2017, 2017, 2756573. [Google Scholar] [CrossRef] [Green Version]
- Schweitzer, H.; Ritter, D.; McIntosh, J.; Barnhart, E.; Cunningham, A.B.; Vinson, D.; Orem, W.; Fields, M.W. Changes in microbial communities and associated water and gas geochemistry across a sulfate gradient in coal beds: Powder River Basin, USA. Geochim. Cosmochim. Acta 2018, 245, 495–513. [Google Scholar] [CrossRef]
- Nobu, M.K.; Narihiro, T.; Kuroda, K.; Mei, R.; Liu, W.-T. Chasing the elusive Euryarchaeota class WSA2: Genomes reveal a uniquely fastidious methyl-reducing methanogen. ISME J. 2016, 10, 2478–2487. [Google Scholar] [CrossRef] [PubMed]
- Chuang, P.-C.; Young, M.B.; Dale, A.W.; Miller, L.G.; Herrera-Silveira, J.A.; Paytan, A. Methane and sulfate dynamics in sediments from mangrove-dominated tropical coastal lagoons, Yucatán, Mexico. Biogeosciences 2016, 13, 2981–3001. [Google Scholar] [CrossRef] [Green Version]
- Oremland, R.S.; Polcin, S. Methanogenesis and Sulfate Reduction: Competitive and Noncompetitive Substrates in Estuarine Sediments. Appl. Environ. Microbiol. 1982, 44, 1270–1276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sela-Adler, M.; Ronen, Z.; Herut, B.; Antler, G.; Vigderovich, H.; Eckert, W.; Sivan, O. Co-existence of Methanogenesis and Sulfate Reduction with Common Substrates in Sulfate-Rich Estuarine Sediments. Front. Microbiol. 2017, 8, 766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, K.-Q.; Beulig, F.; Kjeldsen, K.U.; Jørgensen, B.B.; Risgaard-Petersen, N. Concurrent Methane Production and Oxidation in Surface Sediment from Aarhus Bay, Denmark. Front. Microbiol. 2017, 8, 1198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mcgenity, T.J. Methanogens and methanogenesis in hypersaline environments. In Handbook of Hydrocarbon and Lipid Microbiology, 1st ed.; Timmis, K.N., Ed.; Springer: Berlin/Heidelberg, Germany, 2010; pp. 665–680. [Google Scholar]
- Zhuang, G.-C.; Elling, F.J.; Nigro, L.M.; Samarkin, V.; Joye, S.B.; Teske, A.; Hinrichs, K.-U. Multiple evidence for methylotrophic methanogenesis as the dominant methanogenic pathway in hypersaline sediments from the Orca Basin, Gulf of Mexico. Geochim. Cosmochim. Acta 2016, 187, 1–20. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Sample | Coordinates | Salinity (‰) | Temp. (°C) | D.O. (mg/L) | pH | Average Bacterial Observed ASV | Average Archaeal Observed ASV | Shannon Index |
|---|---|---|---|---|---|---|---|---|
| Area 1 (A1) | 27.364 N, 113.539 W | 60.6 ± 4.72 | 29.5 ± 0.40 | 7.2 | 8.52 ± 0.005 | 644 ± 16 | 61 ± 14 | 12.9 ± 0.1 |
| Area 4 Near Area 1 (A4N1) | 27.601 N, 113.8969 W | 83.3 ± 4.04 | 27.6 ± 0.81 | 6.2 | 8.62 ± 0.005 | 471 ± 25 | 26 ± 5 | 11.1 ± 0.2 |
| Area 4 Near Area 5 (A4N5) | 27.690 N, 113.9210 W | 118.3 ± 2.88 | 26.2 ± 0.46 | 8.0 | 8.32 ± 0.005 | 354 ± 51 | 5 ± 4 | 9.3 ± 0.8 |
| Area 5 (A5) | 27.690 N, 113.9209 W | 123.6 ± 0.57 | 24.7 ± 0.17 | 7.0 | 8.34 ± 0.005 | 413 ± 71 | 36 ± 9 | 10.5 ± 0.8 |
| Gene | E.C. Number | PFAM | Module | Matching Paired Reads | Proportion (%) |
|---|---|---|---|---|---|
| cdhC | EC:2.3.1.169 | PF03598 | acetate => methane | 1585 | 0.0016 |
| pta | EC:2.3.1.8 | PF01515 | acetate => methane | 9999 | 0.0104 |
| ackA | EC:2.7.2.1 | PF00871 | acetate => methane | 10,586 | 0.0110 |
| acs | EC:6.2.1.1 | PF16177 | acetate => methane | 2523 | 0.0026 |
| mtrA-H | EC:2.1.1.86 | PF04208 | acetate => methane | 362 | 0.0004 |
| CO2 => methane | |||||
| mer | EC:1.5.98.2 | PF00296 | CO2 => methane | 3179 | 0.0033 |
| mtd | EC:1.5.98.1 | PF01993 | CO2 => methane | 69 | 7 × 10−5 |
| hmd | EC:1.12.98.2 | PF03201 | CO2 => methane | 83 | 9 × 10−5 |
| mch | EC:3.5.4.27 | PF02289 | CO2 => methane | 864 | 0.0009 |
| ftr | EC:2.3.1.101 | PF01913 | CO2 => methane | 698 | 0.0007 |
| PF02741 | CO2 => methane | 620 | 0.0006 | ||
| fwdA, fmdA | EC:1.2.7.12 | PF01493 | CO2 => methane | 4823 | 0.0050 |
| PF01568 | 4732 | 0.0049 | |||
| mtaA | EC:2.1.1.246 | PF01208 | methanol => methane | 18,823 | 0.0195 |
| mtaB | EC:2.1.1.90 | PF12176 | methanol => methane | 1449 | 0.0015 |
| mtbA | EC:2.1.1.247 | PF01208 | methylamine/dimethylamine/trimethylamine => methane | 18,823 | 0.0195 |
| mttB | EC:2.1.1.250 | PF06253 | methylamine/dimethylamine/trimethylamine => methane | 47,197 | 0.0489 |
| mtbB | EC:2.1.1.249 | PF09505 | methylamine/dimethylamine/trimethylamine => methane | 653 | 0.0007 |
| Dmd | EC:1.5.8.1 | PF00724 | methylamine/dimethylamine/trimethylamine => methane | 7091 | 0.0073 |
| Tmd | EC:1.5.8.2 | PF07992 | methylamine/dimethylamine/trimethylamine => methane | 71,733 | 0.0743 |
| mtmB | EC:2.1.1.248 | PF05369 | methylamine/dimethylamine/trimethylamine => methane | 1750 | 0.0018 |
| hdrA1 | EC:1.8.7.3 | PF00037 | methylamine/dimethylamine/trimethylamine => methane | 56,679 | 0.0587 |
| PF02662 | methanol => methane | 7902 | 0.0082 | ||
| PF07992 | acetate => methane | 71,733 | 0.0743 | ||
| hdrABC | EC:1.8.98.4 | PF00037 | methylamine/dimethylamine/trimethylamine => methane | 56,679 | 0.0587 |
| EC:1.8.98.6 | PF12838 | acetate => methane | 35,515 | 0.0368 | |
| EC:1.8.98.5 | PF07992 | CO2 => methane | 71,733 | 0.0743 | |
| hdrD | EC:1.8.98.1 | PF02754 | methylamine/dimethylamine/trimethylamine => methane | 10,925 | 0.0113 |
| PF13183 | methanol => methane | 13,950 | 0.0145 | ||
| acetate => methane | |||||
| CO2 => methane | |||||
| mcrA | EC:2.8.4.1 | PF02249 | methylamine/dimethylamine/trimethylamine => methane | 139 | 0.0001 |
| PF02745 | methanol => methane | 188 | 0.0002 | ||
| PF02241 | acetate => methane | 166 | 0.0002 | ||
| PF02783 | CO2 => methane | 91 | 9 × 10−5 | ||
| PF02240 | 151 | 0.0002 | |||
| Fhs | EC:6.3.4.3 | PF01268 | C1-unit interconversion; Wood–Ljungdahl pathway | 16,065 | 0.0166 |
| PF00763 | C1-unit interconversion; Wood–Ljungdahl pathway | 3503 | 0.0036 | ||
| PF02882 | C1-unit interconversion; Wood–Ljungdahl pathway | 8293 | 0.0086 | ||
| Fdh | EC:1.17.1.9 | PF04879 | Wood–Ljungdahl pathway | 4417 | 0.0046 |
| PF00384 | Wood–Ljungdahl pathway | 12,428 | 0.0129 | ||
| PF01568 | Wood–Ljungdahl pathway | 4732 | 0.0049 | ||
| cdhA | EC:1.2.7.4 | PF03063 | CO2 => acetyl-CoA; Wood–Ljungdahl pathway | 21,268 | 0.0220 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
García-Maldonado, J.Q.; Latisnere-Barragán, H.; Escobar-Zepeda, A.; Cadena, S.; Ramírez-Arenas, P.J.; Vázquez-Juárez, R.; Rojas-Contreras, M.; López-Cortés, A. Revisiting Microbial Diversity in Hypersaline Microbial Mats from Guerrero Negro for a Better Understanding of Methanogenic Archaeal Communities. Microorganisms 2023, 11, 812. https://doi.org/10.3390/microorganisms11030812
García-Maldonado JQ, Latisnere-Barragán H, Escobar-Zepeda A, Cadena S, Ramírez-Arenas PJ, Vázquez-Juárez R, Rojas-Contreras M, López-Cortés A. Revisiting Microbial Diversity in Hypersaline Microbial Mats from Guerrero Negro for a Better Understanding of Methanogenic Archaeal Communities. Microorganisms. 2023; 11(3):812. https://doi.org/10.3390/microorganisms11030812
Chicago/Turabian StyleGarcía-Maldonado, José Q., Hever Latisnere-Barragán, Alejandra Escobar-Zepeda, Santiago Cadena, Patricia J. Ramírez-Arenas, Ricardo Vázquez-Juárez, Maurilia Rojas-Contreras, and Alejandro López-Cortés. 2023. "Revisiting Microbial Diversity in Hypersaline Microbial Mats from Guerrero Negro for a Better Understanding of Methanogenic Archaeal Communities" Microorganisms 11, no. 3: 812. https://doi.org/10.3390/microorganisms11030812