
Association of Dengue Virus Serotypes 1&2 with Severe Dengue Having Deletions in Their 3′Untranslated Regions (3′UTRs)
Abstract
:1. Introduction
2. Materials and Methods
2.1. Clinical Samples
2.2. Serotyping
2.3. Sequencing and Phylogenetic Analyses
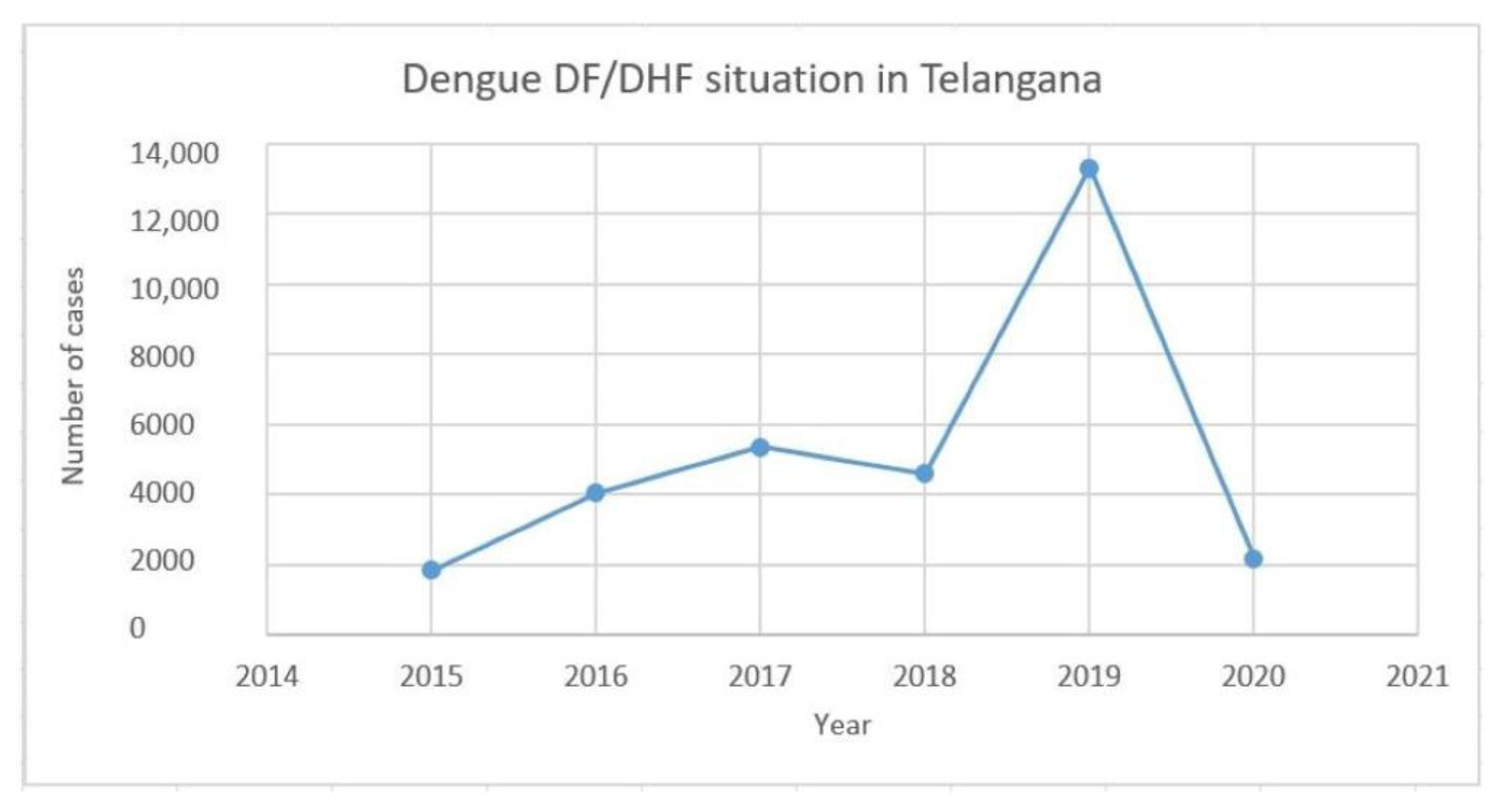
2.4. Epidemiological Data of Dengue Cases in Telangana
2.5. Analysis of 3′ Untranslated Regions (3′UTRs)
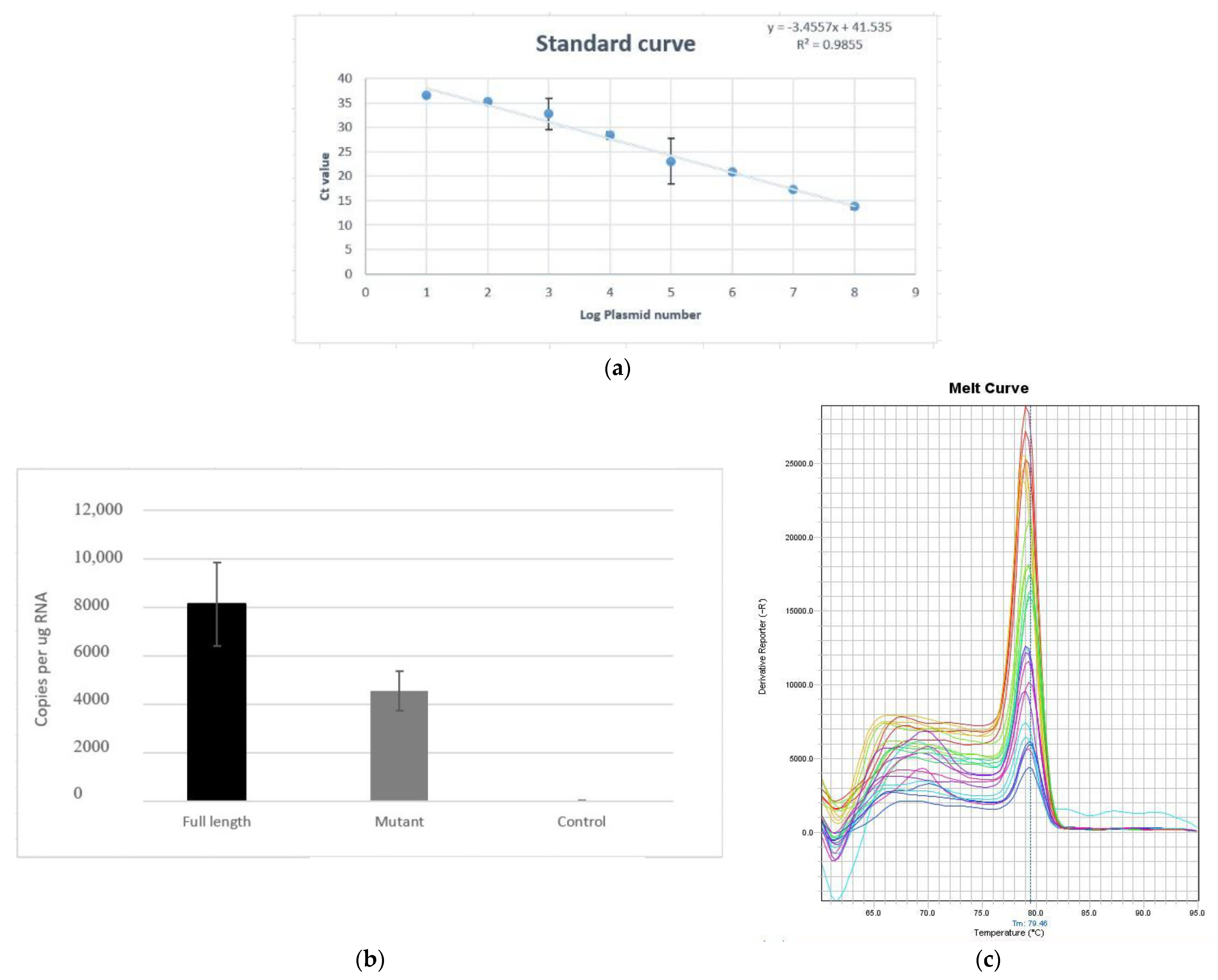
2.6. Analysis of Replication Efficiency of 3′UTR Mutants
2.6.1. Quantitative Real-Time PCR (qPCR)
2.6.2. Structural Analysis of 3′UTRs
3. Results
3.1. Patient Samples
3.2. Analysis of Serotype
3.3. Identification of Genotype
3.4. Analysis of Dengue Cases from 2015 to 2020
3.5. 3′UTR Sequence Analysis
3.6. Viral Copy Number of Full-Length and Mutant 3′UTR Samples
3.7. RNA Structure Analysis of the 3′UTRs
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Acknowledgments
Conflicts of Interest
References
- Bhatt, S.; Gething, P.W.; Brady, O.J.; Messina, J.P.; Farlow, A.W.; Moyes, C.L.; Drake, J.M.; Brownstein, J.S.; Hoen, A.G.; Sankoh, O.; et al. The global distribution and burden of dengue. Nature 2013, 496, 504–507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hadinegoro, S.R.S. The revised WHO dengue case classification: Does the system need to be modified? Paediatr. Int. Child Health 2012, 32, 33–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dwivedi, V.D.; Tripathi, I.P.; Tripathi, R.C.; Bharadwaj, S.; Mishra, S.K. Genomics, proteomics and evolution of dengue virus. Brief. Funct. Genom. 2017, 16, 217–227. [Google Scholar] [CrossRef] [PubMed]
- Leitmeyer, K.C.; Vaughn, D.W.; Watts, D.M.; Salas, R.; Villalobos, I.; Chacon, D.; Ramos, C.; Rico-Hesse, R. Dengue Virus Structural Differences That Correlate with Pathogenesis. J. Virol. 1999, 73, 4738–4747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pollett, S.; Melendrez, M.; Berry, I.M.; Duchêne, S.; Salje, H.; Cummings, D.; Jarman, R. Understanding dengue virus evolution to support epidemic surveillance and counter-measure development. Infect. Genet. Evol. 2018, 62, 279–295. [Google Scholar] [CrossRef] [PubMed]
- Vaddadi, K.; Gandikota, C.; Jain, P.K.; Prasad, V.S.V.; Venkataramana, M. Co-circulation and co-infections of all dengue virus serotypes in Hyderabad, India 2014. Epidemiol. Infect. 2017, 145, 2563–2574. [Google Scholar] [CrossRef] [Green Version]
- Shrivastava, S.; Tiraki, D.; Diwan, A.; Lalwani, S.K.; Modak, M.; Mishra, A.C.; Arankalle, V.A. Co-circulation of all the four dengue virus serotypes and detection of a novel clade of DENV-4 ( genotype I ) virus in Pune, India during 2016 season. PLoS ONE 2018, 13, e0192672. [Google Scholar] [CrossRef] [Green Version]
- Guo, C.; Zhou, Z.; Wen, Z.; Liu, Y.; Zeng, C.; Xiao, D.; Ou, M.; Han, Y.; Huang, S.; Liu, D.; et al. Global epidemiology of dengue outbreaks in 1990–2015: A systematic review and meta-analysis. Front. Cell. Infect. Microbiol. 2017, 7, 317. [Google Scholar] [CrossRef] [Green Version]
- Patil, J.; Alagarasu, K.; Kakade, M.; More, A.; Gadekar, K.; Jadhav, S.; Parashar, D.; Shah, P. Emergence of dengue virus type 1 and type 3 as dominant serotypes during 2017 in Pune and Nashik regions of Maharashtra, Western India. Infect. Genet. Evol. 2018, 66, 272–283. [Google Scholar] [CrossRef]
- Hesse, R.R. Microevolution and virulence of dengue viruses. Adv. Virus Res. 2003, 59, 315–341. [Google Scholar]
- Worobey, M.; Rambaut, A.; Holmes, E.C. Widespread intra-serotype recombination in natural populations of dengue virus. Proc. Natl. Acad. Sci. USA 1999, 96, 7352–7357. [Google Scholar] [CrossRef] [Green Version]
- Mutheneni, S.R.; Mopuri, R.; Naish, S.; Gunti, D.; Upadhyayula, S.M. Spatial distribution and cluster analysis of dengue using self-organizing maps in Andhra Pradesh, India, 2011–2013. Parasite Epidemiol. Control 2018, 3, 52–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gandikota, C.; Mohammed, F.; Gandhi, L.; Maisnam, D.; Mattam, U.; Rathore, D.; Chatterjee, A.; Mallick, K.; Billoria, A.; Prasad, V.S.V.; et al. Mitochondrial Import of Dengue Virus NS3 Protease and Cleavage of GrpEL1, a Cochaperone of Mitochondrial Hsp70. J. Virol. 2021, 94, e01178-20. [Google Scholar] [CrossRef] [PubMed]
- Shu, P.-Y.; Chang, S.-F.; Kuo, Y.-C.; Yueh, Y.-Y.; Chien, L.-J.; Sue, C.-L.; Lin, T.-H.; Huang, J.-H. Development of group- and serotype-specific one-step SYBR Green I-based real-time reverse transcription-PCR assay for dengue virus. J. Clin. Microbiol. 2003, 41, 2408–2416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dash, P.K.; Sharma, S.; Soni, M.; Agarwal, A.; Sahni, A.K.; Parida, M. Complete genome sequencing and evolutionary phylogeography analysis of Indian isolates of Dengue virus type 1. Virus Genes 2015, 195, 124–134. [Google Scholar] [CrossRef]
- Guzman, M.G.; Halstead, S.B.; Artsob, H.; Buchy, P.; Farrar, J.; Gubler, D.J.; Hunsperger, E.; Kroeger, A.; Margolis, H.S.; Martínez, E.; et al. Dengue: A continuing global threat. Nat. Rev. Microbiol. 2010, 8, S7–S16. [Google Scholar] [CrossRef] [Green Version]
- Kukreti, H.; Dash, P.K.; Parida, M.; Chaudhary, A.; Saxena, P.; Rautela, R.; Mittal, V.; Chhabra, M.; Bhattacharya, D.; Lal, S.; et al. Phylogenetic studies reveal existence of multiple lineages of a single genotype of DENV-1 (genotype III) in India during 1956–2007. Virol. J. 2009, 6. [Google Scholar] [CrossRef] [Green Version]
- Ahamed, S.F.; Rosario, V.; Britto, C.; Dias, M.; Nayak, K.; Chandele, A.; Kaja, M.-K.; Shet, A. Emergence of new genotypes and lineages of dengue viruses during the 2012–15 epidemics in southern India. Int. J. Infect. Dis. 2019, 84, S34–S43. [Google Scholar] [CrossRef] [Green Version]
- Murugesan, A.; Aridoss, D.; Senthilkumar, S.; Sivathanu, L.; Sekar, R.; Shankar, E.M.; Manickan, E. Molecular diversity of dengue virus serotypes 1-4 during an outbreak of acute dengue virus infection in Theni, India. Indian J. Med. Microbiol. 2020, 38, 401–408. [Google Scholar] [CrossRef]
- Neeraja, M.; Lakshmi, V.; Lavanya, V.; Priyanka, E.; Parida, M.; Dash, P.; Sharma, S.; Rao, P.L.; Reddy, G. Rapid detection and differentiation of dengue virus serotypes by NS1 specific reverse transcription loop-mediated isothermal amplification(RT-Lamp) assay in patients presenting to a tertiary care hospital in hyderabad, India. J. Virol. Methods 2015, 211, 22–31. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Anish, T.S.; Valamparampil, M.J.; Thomas, A.T.; Mathew, A.J.; Ajithlal, P.M.; Jambulingam, P. Genotype shift of dengue virus (DENV1) during the 2017 outbreak of dengue fever in Thiruvananthapuram, Kerala, India. Indian J. Exp. Biol. 2019, 57, 961–966. [Google Scholar]
- Pooja, S.; Sabeena, S.; Revti, B.; Sanjay, R.; Anjali, A.; Rajendra, K.; Aswathyraj, S.; Giselle, D.; Hindol, M.; Arunkumar, G. Circulating genotypes of dengue-1 virus in South West India, 2014–2015. Jpn. J. Infect. Dis. 2017, 70, 663–665. [Google Scholar] [CrossRef] [Green Version]
- Chen, R.; Vasilakis, N. Dengue-Quo tu et quo vadis? Viruses 2011, 3, 1562–1608. [Google Scholar] [CrossRef] [PubMed]
- Tajima, S.; Nukui, Y.; Takasaki, T.; Kurane, I.; Jung, J.K.; Kwun, H.J.; Lee, J.-O.; Arora, P.; Jang, K.L. Characterization of the variable region in the 3’ non-translated region of dengue type 1 virus. J. Gen. Virol. 2007, 88, 2214–2222. [Google Scholar] [CrossRef] [PubMed]
- OiAinle, M.; Balmaseda, A.; Macalalad, A.R.; Tellez, Y.; Zody, M.C.; Saborio, S.; Nunez, A.; Lennon, N.J.; Birren, B.W.; Gordon, A.; et al. Dynamics of Dengue Disease Severity Determined by the Interplay Between Viral Genetics and Serotype-Specific Immunity. Sci. Transl. Med. 2011, 3, 114–128. [Google Scholar]
- Barde, P.; Shukla, M.; Kori, B.; Chand, G.; Jain, L.; Varun, B.; Dutta, D.; Baruah, K.; Singh, N. Emergence of dengue in tribal villages of Mandla district, Madhya Pradesh, India. Indian J. Med. Res. 2015, 141, 584–590. [Google Scholar]
- Dayananda, P.D.; de Silva, H.; Fernando, L.; de Silva, B.G.D.N.K. Genetic Variation in the Domain II, 3’ Untranslated Region of Human and Mosquito Derived Dengue Virus Strains in Sri Lanka. Viruses 2021, 13, 421. [Google Scholar] [CrossRef]
- Pankhong, P.; Weiner, D.B.; Ramanathan, M.P.; Nisalak, A.; Kalayanarooj, S.; Nimmanithya, S.; Attatippaholkun, W. Molecular genetic relationship of the 3’Untranslated region among Thai Dengue-3 virus, Bangkok isolates, During 1973-2000. DNA Cell Biol. 2009, 28, 481–491. [Google Scholar] [CrossRef] [PubMed]
- Tajima, S.; Nukui, Y.; Ito, M.; Takasaki, T.; Kurane, I. Nineteen nucleotides in the variable region of 3’non-translated region is dispensable for the replication of dengue type 1 virus in vitro. Virus Res. 2005, 116, 38–44. [Google Scholar] [CrossRef]
- Blaney, J.E.; Sathe, N.S.; Goddard, L.; Hanson, C.T.; Romero, T.A.; Hanley, K.A.; Murphy, B.R.; Whitehead, S.S. Dengue virus type 3 vaccine candidates generated by introduction of deletions in the 3 untranslated region (3-UTR) or by exchange of the DENV-3 3’UTRwith that of DENV-4. Vaccine 2008, 26, 817–828. [Google Scholar] [CrossRef] [Green Version]
- Islam, A.; Abdullah, M.; Tazeen, A.; Naqvi, I.H.; Kazim, S.N.; Ahmed, A.; Alamery, S.F.; Malik, A.; Parveen, S. Circulation of dengue virus serotypes in hyperendemic region of New Delhi, India during 2011–2017. J. Infect. Public Health 2020, 13, 1912–1919. [Google Scholar] [CrossRef]
- Srikiatkhachorn, A.; Green, S. Markers of Dengue Disease Severity. Curr. Top. Microbiol. Immunol. 2010, 338, 67–82. [Google Scholar]
- Chaturvedi, U.C.; Nagar, R. Dengue and dengue haemorrhagic fever: Indian perspective. J. Biosci. 2008, 33, 429–441. [Google Scholar] [CrossRef] [PubMed]
- Betancur, J.C.; Inchima, S.C. Overexpression of miR-484 and miR-744 in Vero cells alters Dengue virus replication. Mem. Inst. Oswaldo Cruz. 2017, 112, 281–291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castillo, J.A.; Castrillon, J.C.; Toro, M.D.; Bentancur, J.C.; St Lauren, G., III; Smit, J.M.; Urcuqui-Inchima, S. Complex interaction between dengue virus replication and expression of miRNA-133a. BMC Infect. Dis. 2016, 16, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tambyah, P.A.; Ching, C.S.; Sepramaniam, S.; Ali, J.M.; Armugam, A.; Jeeyasalin, K. microRNA expression in blood of dengue patients. Ann. Clin. Biochem. 2016, 53, 466–476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skalsky, R.L.; Cullen, B.R. Viruses, microRNAs, and Host Interactions. Annu. Rev. Microbiol. 2010, 64, 123–141. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Xie, J.; Xu, X.; Wang, J.; Ao, F.; Wan, Y.; Zhu, Y. MicroRNA-548 down-regulates host antiviral response via direct targeting of IFN-λ1. Protein Cell 2013, 4, 130–141. [Google Scholar] [CrossRef] [Green Version]
- Pedersen, I.M.; Cheng, G.; Wieland, S.; Volinia, S.; Croce, C.M.; Chisari, F.V.; David, M. Interferon modulation of cellular microRNAs as an antiviral mechanism. Nature 2007, 449, 919–922. [Google Scholar] [CrossRef] [Green Version]
- Wu, S.; He, L.; Li, Y.; Wang, T.; Feng, L.; Jiang, L.; Zhang, P.; Huang, X. miR-146a facilitates replication of dengue virus by dampening interferon induction by targeting TRAF6. J. Infect. 2013, 67, 329–341. [Google Scholar] [CrossRef]


 symbol, which are clustered in GI (Asian genotype). Previous GIII (sylvatic strain) is also indicated by the
symbol, which are clustered in GI (Asian genotype). Previous GIII (sylvatic strain) is also indicated by the  symbol. The sequences are indicated by their accession number, country, and year of collection.
symbol, which are clustered in GI (Asian genotype). Previous GIII (sylvatic strain) is also indicated by the symbol. The sequences are indicated by their accession number, country, and year of collection.
symbol. The sequences are indicated by their accession number, country, and year of collection.
symbol, which are clustered in GI (Asian genotype). Previous GIII (sylvatic strain) is also indicated by the symbol. The sequences are indicated by their accession number, country, and year of collection.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Serotype | Accession No. | Type of Dengue | No. of Deleted Nucleotides | Age/Sex | Thrombocytopenia | Pleural Effusion | Abdominal Pain | Hematomegaly | NS1/IgM/IgG | AST/ALT |
|---|---|---|---|---|---|---|---|---|---|---|
| DENV-1 | KX618705 | DF | Full length | 8 Y/M | Yes | No | No | No | +/−/− | Mild |
| DENV-1 | KX618706 | DHF | Full length | 12 Y/F | Yes | No | Yes | Mild | +/−/− | Normal |
| a DENV-1 | MG560149 | DHF | 22 | 8 M/M | Yes | Yes | Yes | Yes | +/−/− | Normal |
| a DENV-1 | OM572555 | Dengue with warning signs | 22 | 2 Y/M | No | No | No | No | +/+/− | Normal |
| a DENV-1 | OM572558 | Severe dengue with myocarditis | 22 | 4 Y/F | Mild | Yes | Yes | Yes | +/+/− | Mild |
| DENV-1 | MG560150 | DF | 8 | 3 Y/F | No | No | No | Yes | +/−/− | Normal |
| DENV-1 | MG560151 | DF | 8 | 11 Y/M | Yes | No | Yes | No | +/−/− | Elevated |
| a DENV-2 | MG560144 | Severe dengue | 50 | 13 Y/M | Yes | Mild | Yes | Yes | +/−/− | Elevated |
| DENV-2 | MG560143 | Severe dengue with Septic shock | Full length | 7 Y/F | Yes | Yes | Yes | Yes | +/+/− | Elevated |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maisnam, D.; Billoria, A.; Prasad, V.S.V.; Venkataramana, M. Association of Dengue Virus Serotypes 1&2 with Severe Dengue Having Deletions in Their 3′Untranslated Regions (3′UTRs). Microorganisms 2023, 11, 666. https://doi.org/10.3390/microorganisms11030666
Maisnam D, Billoria A, Prasad VSV, Venkataramana M. Association of Dengue Virus Serotypes 1&2 with Severe Dengue Having Deletions in Their 3′Untranslated Regions (3′UTRs). Microorganisms. 2023; 11(3):666. https://doi.org/10.3390/microorganisms11030666
Chicago/Turabian StyleMaisnam, Deepti, Arcy Billoria, V. S. V Prasad, and Musturi Venkataramana. 2023. "Association of Dengue Virus Serotypes 1&2 with Severe Dengue Having Deletions in Their 3′Untranslated Regions (3′UTRs)" Microorganisms 11, no. 3: 666. https://doi.org/10.3390/microorganisms11030666