

Changes of In Situ Prokaryotic and Eukaryotic Communities in the Upper Sanya River to the Sea over a Nine-Hour Period
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. DNA Extraction and Sequencing
2.3. Microbial Community Analysis
2.4. Prokaryotic and Eukaryotic Microbial Community Assembly Predicted by Neutral Community Model
3. Results and Discussion
3.1. Sequencing Statistics and Microbial Diversity
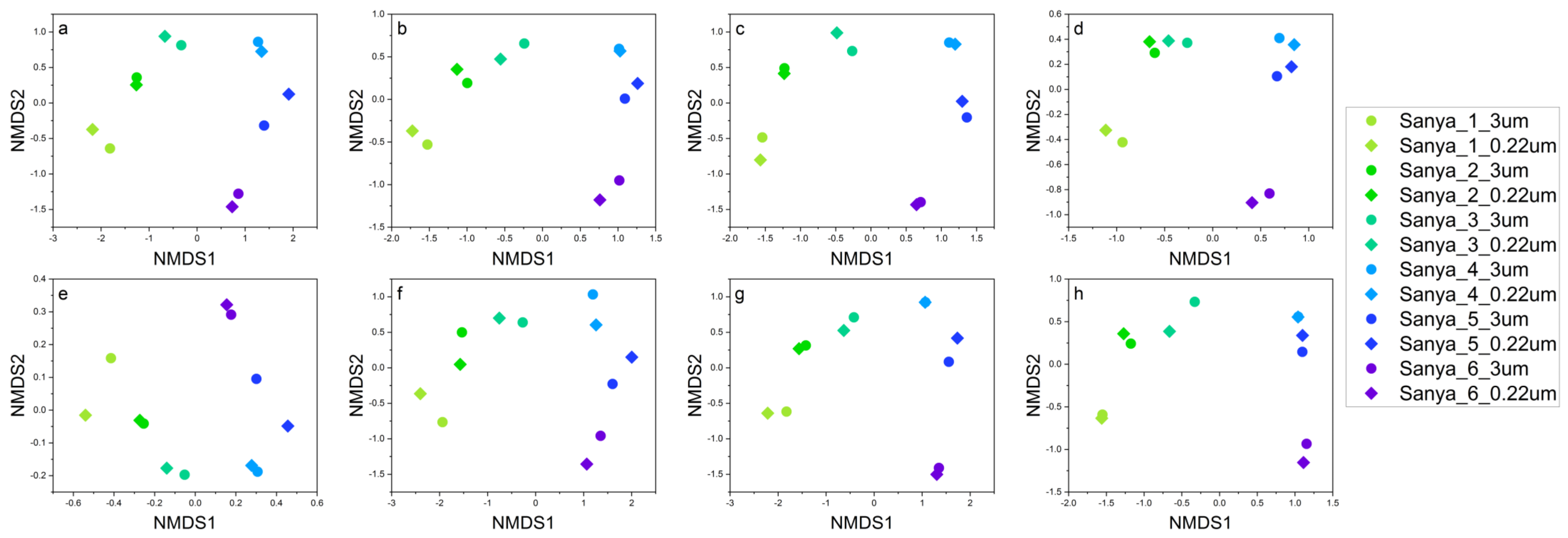
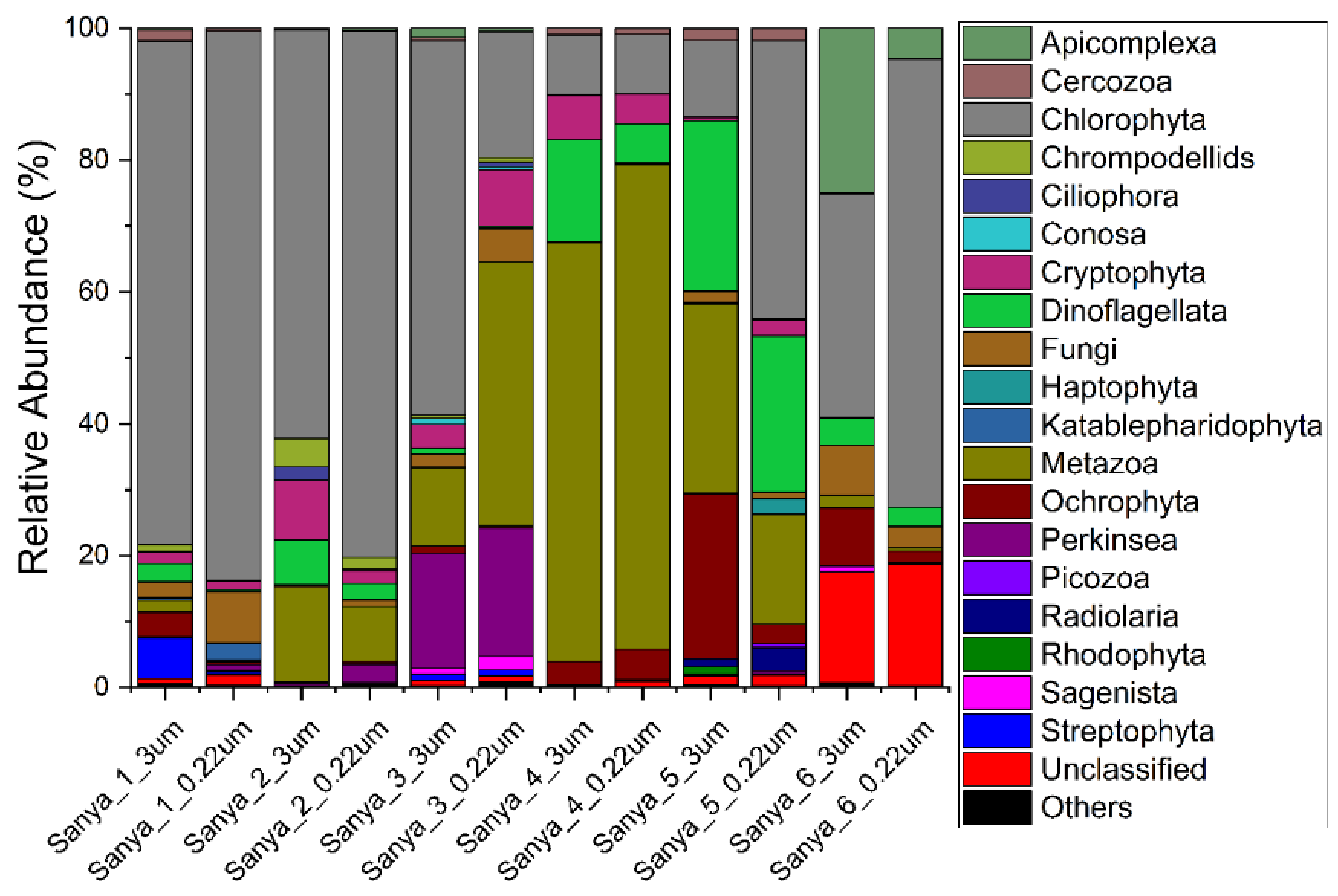
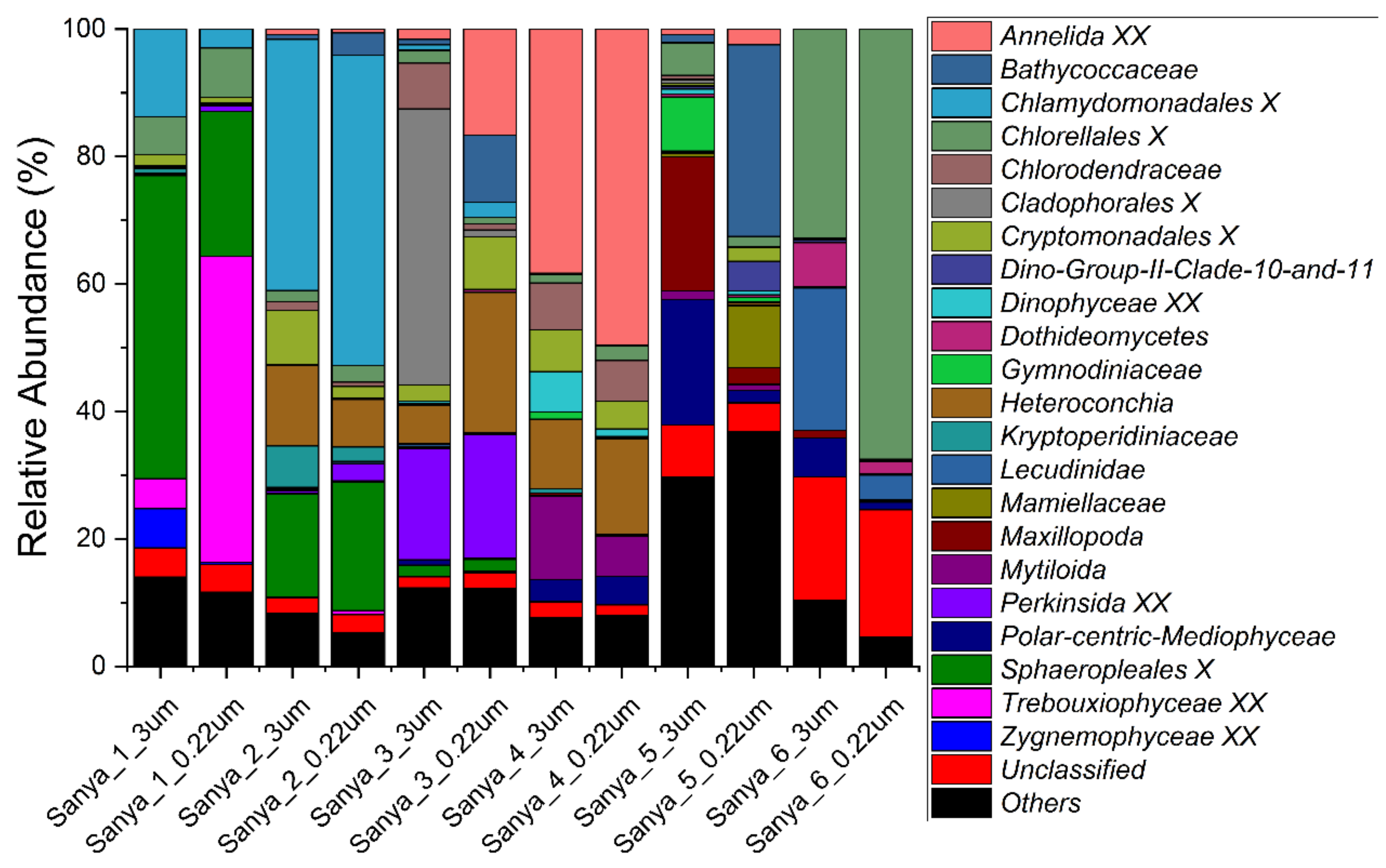
3.2. Structure and Composition of the Microbial Communities
3.3. The Shared and Unique Prokaryotic and Eukaryotic Lineages of Samples Collected from Different Sampling Sites
3.4. The Neutral Model of Ccommunity Assembly Based on Abundant, Common, and Rare Microbial Datasets
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Prosser, J.I.; Bohannan, B.J.; Curtis, T.P.; Ellis, R.J.; Firestone, M.K.; Freckleton, R.P.; Green, J.L.; Green, L.E.; Killham, K.; Lennon, J.J.; et al. The role of ecological theory in microbial ecology. Nat. Rev. Microbiol. 2007, 5, 384–392. [Google Scholar] [CrossRef] [PubMed]
- Falkowski, P.G.; Fenchel, T.; Delong, E.F. The microbial engines that drive Earth’s biogeochemical cycles. Science 2008, 320, 1034–1039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marron, A.O.; Ratcliffe, S.; Wheeler, G.L.; Goldstein, R.E.; King, N.; Not, F.; de Vargas, C.; Richter, D.J. The Evolution of Silicon Transport in Eukaryotes. Mol. Biol. Evol. 2016, 33, 3226–3248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hutchins, D.A.; Fu, F. Microorganisms and ocean global change. Nat. Microbiol. 2017, 2, 17058. [Google Scholar] [CrossRef]
- Tran, P.Q.; Anantharaman, K. Biogeochemistry Goes Viral: Towards a Multifaceted Approach To Study Viruses and Biogeochemical Cycling. mSystems 2021, 6, e0113821. [Google Scholar] [CrossRef]
- Ensign, S.H.; Doyle, M.W. Nutrient spiraling in streams and river networks. J. Geophys. Res. Biogeosci. 2006, 111, G04009. [Google Scholar] [CrossRef]
- Cole, J.J.; Prairie, Y.T.; Caraco, N.F.; McDowell, W.H.; Tranvik, L.J.; Striegl, R.G.; Duarte, C.M.; Kortelainen, P.; Downing, J.A.; Middelburg, J.J.; et al. Plumbing the Global Carbon Cycle: Integrating Inland Waters into the Terrestrial Carbon Budget. Ecosystems 2007, 10, 172–185. [Google Scholar] [CrossRef] [Green Version]
- Withers, P.J.; Jarvie, H.P. Delivery and cycling of phosphorus in rivers: A review. Sci. Total Environ. 2008, 400, 379–395. [Google Scholar] [CrossRef]
- Battin, T.J.; Luyssaert, S.; Kaplan, L.A.; Aufdenkampe, A.K.; Richter, A.; Tranvik, L.J. The boundless carbon cycle. Nature Geosci. 2009, 2, 598–600. [Google Scholar] [CrossRef]
- Read, D.S.; Gweon, H.S.; Bowes, M.J.; Newbold, L.K.; Field, D.; Bailey, M.J.; Griffiths, R.I. Catchment-scale biogeography of riverine bacterioplankton. ISME J. 2015, 9, 516–526. [Google Scholar] [CrossRef] [Green Version]
- Cruaud, P.; Vigneron, A.; Fradette, M.S.; Dorea, C.C.; Culley, A.I.; Rodriguez, M.J.; Charette, S.J. Annual bacterial community cycle in a seasonally ice-covered river reflects environmental and climatic conditions. Limnol. Oceanogr. 2020, 65, S21–S37. [Google Scholar] [CrossRef]
- Liu, T.; Zhang, A.N.; Wang, J.; Liu, S.; Jiang, X.; Dang, C.; Ma, T.; Liu, S.; Chen, Q.; Xie, S.; et al. Integrated biogeography of planktonic and sedimentary bacterial communities in the Yangtze River. Microbiome 2018, 6, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Savio, D.; Sinclair, L.; Ijaz, U.Z.; Parajka, J.; Reischer, G.H.; Stadler, P.; Blaschke, A.P.; Blöschl, G.; Mach, R.L.; Kirschner, A.K.; et al. Bacterial diversity along a 2600 km river continuum. Environ. Microbiol. 2015, 17, 4994–5007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muneepeerakul, R.; Bertuzzo, E.; Lynch, H.J.; Fagan, W.F.; Rinaldo, A.; Rodriguez-Iturbe, I. Neutral metacommunity models predict fish diversity patterns in Mississippi-Missouri basin. Nature 2008, 453, 220–222. [Google Scholar] [CrossRef] [Green Version]
- Blackman, R.C.; Ho, H.C.; Walser, J.C.; Altermatt, F. Spatio-temporal patterns of multi-trophic biodiversity and food-web characteristics uncovered across a river catchment using environmental DNA. Commun. Biol. 2022, 5, 259. [Google Scholar] [CrossRef]
- Altermatt, F.; Seymour, M.; Martinez, N. River network properties shape alpha-diversity and community similarity patterns of aquatic insect communities across major drainage basins. J. Biogeogr. 2013, 40, 2249–2260. [Google Scholar] [CrossRef]
- Besemer, K.; Singer, G.; Quince, C.; Bertuzzo, E.; Sloan, W.; Battin, T.J. Headwaters are critical reservoirs of microbial diversity for fluvial networks. Proc. Biol. Sci. 2013, 280, 20131760. [Google Scholar] [CrossRef]
- Ficetola, G.F.; Miaud, C.; Pompanon, F.; Taberlet, P. Species detection using environmental DNA from water samples. Biol. Lett. 2008, 4, 423–425. [Google Scholar] [CrossRef] [Green Version]
- Barnes, M.A.; Turner, C.R. The ecology of environmental DNA and implications for conservation genetics. Conserv. Genet. 2016, 17, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Deiner, K.; Altermatt, F. Transport distance of invertebrate environmental DNA in a natural river. PLoS ONE 2014, 9, e88786. [Google Scholar] [CrossRef] [Green Version]
- Korajkic, A.; Parfrey, L.W.; McMinn, B.R.; Baeza, Y.V.; VanTeuren, W.; Knight, R.; Shanks, O.C. Changes in bacterial and eukaryotic communities during sewage decomposition in Mississippi river water. Water Res. 2015, 69, 30–39. [Google Scholar] [CrossRef]
- Liu, L.; Wang, S.; Chen, J. Transformations from specialists to generalists cause bacterial communities are more stable than micro-eukaryotic communities under anthropogenic activity disturbance. Sci. Total Environ. 2021, 790, 148141. [Google Scholar] [CrossRef]
- Yang, J.; Lv, J.; Liu, Q.; Nan, F.; Li, B.; Xie, S.; Feng, J. Seasonal and spatial patterns of eukaryotic phytoplankton communities in an urban river based on marker gene. Sci. Rep. 2021, 11, 23147. [Google Scholar] [CrossRef]
- Ge, Y.; Lou, Y.; Xu, M.; Wu, C.; Meng, J.; Shi, L.; Xia, F.; Xu, Y. Spatial distribution and influencing factors on the variation of bacterial communities in an urban river sediment. Environ. Pollut. 2021, 272, 115984. [Google Scholar] [CrossRef]
- Gao, F.Z.; He, L.Y.; Hu, L.X.; Chen, J.; Yang, Y.Y.; Zou, H.Y.; He, L.X.; Bai, H.; Liu, Y.S.; Zhao, J.L.; et al. Anthropogenic activities and seasonal properties jointly drive the assemblage of bacterial communities in subtropical river basins. Sci. Total Environ. 2022, 806, 151476. [Google Scholar] [CrossRef]
- Nemergut, D.R.; Schmidt, S.K.; Fukami, T.; O’Neill, S.P.; Bilinski, T.M.; Stanish, L.F.; Knelman, J.E.; Darcy, J.L.; Lynch, R.C.; Wickey, P.; et al. Patterns and processes of microbial community assembly. Microbiol. Mol. Biol. Rev. 2013, 77, 342–356. [Google Scholar] [CrossRef] [Green Version]
- Vannote, R.L.; Minshall, G.W.; Cummins, K.W.; Sedell, J.R.; Cushing, C.E. The River Continuum Concept. Can. J. Fish. Aquat. Sci. 1980, 37, 130–137. [Google Scholar] [CrossRef]
- Roguet, A.; Newton, R.J.; Eren, A.M.; McLellan, S.L. Guts of the Urban Ecosystem: Microbial Ecology of Sewer Infrastructure. mSystems 2022, 7, e0011822. [Google Scholar] [CrossRef] [PubMed]
- Niño-García, J.P.; Ruiz-González, C.; Del Giorgio, P.A. Interactions between hydrology and water chemistry shape bacterioplankton biogeography across boreal freshwater networks. ISME J. 2016, 10, 1755–1766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeLeon-Rodriguez, N.; Lathem, T.L.; Rodriguez-R, L.M.; Barazesh, J.M.; Anderson, B.E.; Beyersdorf, A.J.; Ziemba, L.D.; Bergin, M.; Nenes, A.; Konstantinidis, K.T. Microbiome of the upper troposphere: Species composition and prevalence, effects of tropical storms, and atmospheric implications. Proc. Natl. Acad. Sci. USA 2013, 110, 2575–2580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jensen, L.Z.; Glasius, M.; Gryning, S.E.; Massling, A.; Finster, K.; Šantl-Temkiv, T. Seasonal Variation of the Atmospheric Bacterial Community in the Greenlandic High Arctic Is Influenced by Weather Events and Local and Distant Sources. Front. Microbiol. 2022, 13, 909980. [Google Scholar] [CrossRef]
- Crump, R.C.; Adams, H.E.; Hobbie, J.E.; Kling, G.W. Biogeography of bacterioplankton in lakes and streams of an Arctic tundra catchment. Ecology 2007, 88, 1365–1378. [Google Scholar] [CrossRef] [Green Version]
- Crump, B.C.; Amaral-Zettler, L.A.; Kling, G.W. Microbial diversity in arctic freshwaters is structured by inoculation of microbes from soils. ISME J. 2012, 6, 1629–1639. [Google Scholar] [CrossRef] [Green Version]
- Ruiz-González, C.; Niño-García, J.P.; Del Giorgio, P.A. Terrestrial origin of bacterial communities in complex boreal freshwater networks. Ecol. Lett. 2015, 18, 1198–1206. [Google Scholar] [CrossRef]
- Logares, R.; Bråte, J.; Bertilsson, S.; Clasen, J.L.; Shalchian-Tabrizi, K.; Rengefors, K. Infrequent marine-freshwater transitions in the microbial world. Trends Microbiol. 2009, 17, 414–422. [Google Scholar] [CrossRef] [PubMed]
- Pedrós-Alió, C. The rare bacterial biosphere. Ann. Rev. Mar. Sci. 2012, 4, 449–466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lynch, M.D.; Neufeld, J.D. Ecology and exploration of the rare biosphere. Nat. Rev. Microbiol. 2015, 13, 217–229. [Google Scholar] [CrossRef] [PubMed]
- Jiao, S.; Luo, Y.; Lu, M.; Xiao, X.; Lin, Y.; Chen, W.; Wei, G. Distinct succession patterns of abundant and rare bacteria in temporal microcosms with pollutants. Environ. Pollut. 2017, 225, 497–505. [Google Scholar] [CrossRef]
- Teira, E.; Lekunberri, I.; Gasol, J.M.; Nieto-Cid, M.; Alvarez-Salgado, X.A.; Figueiras, F.G. Dynamics of the hydrocarbon-degrading Cycloclasticus bacteria during mesocosm-simulated oil spills. Environ. Microbiol. 2007, 9, 2551–2562. [Google Scholar] [CrossRef] [Green Version]
- Campbell, B.J.; Yu, L.; Heidelberg, J.F.; Kirchman, D.L. Activity of abundant and rare bacteria in a coastal ocean. Proc. Natl. Acad. Sci. USA 2011, 108, 12776–12781. [Google Scholar] [CrossRef] [Green Version]
- Walters, W.; Hyde, E.R.; Berg-Lyons, D.; Ackermann, G.; Humphrey, G.; Parada, A.; Gilbert, J.A.; Jansson, J.K.; Caporaso, J.G.; Fuhrman, J.A.; et al. Improved Bacterial 16S rRNA Gene (V4 and V4-5) and Fungal Internal Transcribed Spacer Marker Gene Primers for Microbial Community Surveys. mSystems 2015, 1, e00009–e00015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bråte, J.; Logares, R.; Berney, C.; Ree, D.K.; Klaveness, D.; Jakobsen, K.S.; Shalchian-Tabrizi, K. Freshwater Perkinsea and marine-freshwater colonizations revealed by pyrosequencing and phylogeny of environmental rDNA. ISME J. 2010, 4, 1144–1153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magoč, T.; Salzberg, S.L. FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef] [Green Version]
- Kong, Y. Btrim: A fast, lightweight adapter and quality trimming program for next-generation sequencing technologies. Genomics 2011, 98, 152–153. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.C. UNOISE2: Improved error-correction for Illumina 16S and ITS amplicon sequencing. BioRxiv 2016, 081257. [Google Scholar] [CrossRef] [Green Version]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glockner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids. Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef] [PubMed]
- Guillou, L.; Bachar, D.; Audic, S.; Bass, D.; Berney, C.; Bittner, L.; Boutte, C.; Burgaud, G.; de Vargas, C.; Decelle, J.; et al. The Protist Ribosomal Reference database (PR2): A catalog of unicellular eukaryote small sub-unit rRNA sequences with curated taxonomy. Nucleic Acids Res. 2013, 41, D597–D604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, L.; Yang, J.; Yu, Z.; Wilkinson, D.M. The biogeography of abundant and rare bacterioplankton in the lakes and reservoirs of China. ISME J. 2015, 9, 2068–2077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Hou, F.; Xie, W.; Wang, K.; Zhou, X.; Zhang, D.; Zhu, X. Interaction and assembly processes of abundant and rare microbial communities during a diatom bloom process. Environ. Microbiol. 2020, 22, 1707–1719. [Google Scholar] [CrossRef]
- Chao, A. Nonparametric-Estimation of the Number of Classes in a Population. Scandinavian. J. Stat. 1984, 11, 265–270. [Google Scholar]
- R Core Team. R: A Language and Environment for Statistical Computing; The R Foundation for Statistical Computing: Vienna, Austria, 2022; Available online: http://www.R-project.org/ (accessed on 27 December 2022).
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J.; et al. Introducing mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef] [Green Version]
- Hubbell, S.P. The Unified Neutral Theory of Biodiversity and Biogeogrpahy; Princton University Press: Princeton, NJ, USA, 2001. [Google Scholar]
- Sloan, W.T.; Lunn, M.; Woodcock, S.; Head, I.M.; Nee, S.; Curtis, T.P. Quantifying the roles of immigration and chance in shaping prokaryote community structure. Environ. Microbiol. 2006, 8, 732–740. [Google Scholar] [CrossRef]
- Chen, W.; Ren, K.; Isabwe, A.; Chen, H.; Liu, M.; Yang, J. Stochastic processes shape microeukaryotic community assembly in a subtropical river across wet and dry seasons. Microbiome 2019, 7, 138. [Google Scholar] [CrossRef] [Green Version]
- Harrell, F.E., Jr.; Dupont, M.C. R Package ‘Hmisc’; R Foundation for Statistical Computing: Vienna, Austria, 2018. [Google Scholar]
- Brown, B.L.; Swan, C.M.; Auerbach, D.A.; Campbell Grant, E.H.; Hitt, N.P.; Maloney, K.O.; Patrick, C. Metacommunity theory as a multispecies, multiscale framework for studying the influence of river network structure on riverine communities and ecosystems. J. North Am. Benthological Soc. 2011, 30, 310–327. [Google Scholar] [CrossRef]
- Madsen, E.L. Microorganisms and their roles in fundamental biogeochemical cycles. Curr. Opin. Biotechnol. 2011, 22, 456–464. [Google Scholar] [CrossRef]
- Prosser, J.I. Ecosystem processes and interactions in a morass of diversity. FEMS Microbiol. Ecol. 2012, 81, 507–519. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Soininen, J.; Han, B.P.; Declerck, S.A.J.; Whittaker, R. Effects of connectivity, dispersal directionality and functional traits on the metacommunity structure of river benthic diatoms. J. Biogeogr. 2013, 40, 2238–2248. [Google Scholar] [CrossRef]
- Banerjee, S.; Kirkby, C.A.; Schmutter, D.; Bissett, A.; Kirkegaard, J.A.; Richardson, A.E. Network analysis reveals functional redundancy and keystone taxa amongst bacterial and fungal communities during organic matter decomposition in an arable soil. Soil Biol. Biochem. 2016, 97, 188–198. [Google Scholar] [CrossRef]
- Liu, S.H.; Zeng, G.M.; Niu, Q.Y.; Liu, Y.; Zhou, L.; Jiang, L.H.; Tan, X.F.; Xu, P.; Zhang, C.; Cheng, M. Bioremediation mechanisms of combined pollution of PAHs and heavy metals by bacteria and fungi: A mini review. Bioresour. Technol. 2017, 224, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Ning, D. Stochastic Community Assembly: Does It Matter in Microbial Ecology? Microbiol. Mol. Biol. Rev. 2017, 81, e00002–e00017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ntougias, S.; Polkowska, Ż.; Nikolaki, S.; Dionyssopoulou, E.; Stathopoulou, P.; Doudoumis, V.; Ruman, M.; Kozak, K.; Namieśnik, J.; Tsiamis, G. Bacterial Community Structures in Freshwater Polar Environments of Svalbard. Microbes. Environ. 2016, 31, 401–409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Cheng, Y.; Gao, G.; Jiang, J. Spatial-Temporal Variation of Bacterial Communities in Sediments in Lake Chaohu, a Large, Shallow Eutrophic Lake in China. Int. J. Environ. Res. Public Health 2019, 16, 3966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jørgensen, N.O.G.; Brandt, K.K.; Nybroe, O.; Hansen, M. Vogesella mureinivorans sp. nov., a peptidoglycan-degrading bacterium from lake water. Int. J. Syst. Evol. Microbiol. 2010, 60, 2467–2472. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.M.; Chen, J.C.; Wang, C.; Huang, C.W.; Sheu, S.Y. Vogesella amnigena sp. nov., isolated from a freshwater river. Int. J. Syst. Evol. Microbiol. 2015, 65, 3634–3640. [Google Scholar] [CrossRef] [PubMed]
- Hoarfrost, A.; Nayfach, S.; Ladau, J.; Yooseph, S.; Arnosti, C.; Dupont, C.L.; Pollard, K.S. Global ecotypes in the ubiquitous marine clade SAR86. ISME J. 2020, 14, 178–188. [Google Scholar] [CrossRef] [Green Version]
- Dinasquet, J.; Landa, M.; Obernosterer, I. SAR11 clade microdiversity and activity during the early spring blooms off Kerguelen Island, Southern Ocean. Environ. Microbiol. Rep. 2022, 14, 907–916. [Google Scholar] [CrossRef]
- Gao, P.; Du, G.; Zhao, D.; Wei, Q.; Zhang, X.; Qu, L.; Gong, X. Influences of Seasonal Monsoons on the Taxonomic Composition and Diversity of Bacterial Community in the Eastern Tropical Indian Ocean. Front. Microbiol. 2021, 11, 615221. [Google Scholar] [CrossRef]
- Reintjes, G.; Tegetmeyer, H.E.; Bürgisser, M.; Orlić, S.; Tews, I.; Zubkov, M.; Voß, D.; Zielinski, O.; Quast, C.; Glöckner, F.O.; et al. On-Site Analysis of Bacterial Communities of the Ultraoligotrophic South Pacific Gyre. Appl. Environ. Microbiol. 2019, 85, e00184-19. [Google Scholar] [CrossRef] [Green Version]
- Benson, A.A. Following the path of carbon in photosynthesis: A personal story. Photosynth. Res. 2002, 73, 29–49. [Google Scholar] [CrossRef]
- Kotut, K.; Krienitz, L.; Scheffler, W.; Engels, M.; Grigorszky, I. Some interesting cyanoprokaryotes and algae from Turkwel Gorge Reservoir, Kenya. Algological. Studies 1998, 91, 37–55. [Google Scholar]
- Massalski, A.; Mrozinska, T.; Olech, M. Ultrastructure of Lobosphaera reniformis (Watanabe) Komárek et Fott (Chlorellales) from King George Island, South Shetland Islands, Antarctica. Acta Societatis Botanicorum. Poloniae 1994, 63, 205–210. [Google Scholar] [CrossRef] [Green Version]
- Gattás, F.; De Stefano, L.G.; Vinocur, A.; Bordet, F.; Espinosa, M.S.; Pizarro, H.; Cataldo, D. Impact of interaction between Limnoperna fortunei and Roundup Max® on freshwater phytoplankton: An in situ approach in Salto Grande reservoir (Argentina). Chemosphere 2018, 209, 748–757. [Google Scholar] [CrossRef] [Green Version]
- Miyasaka, H.; Tanaka, S.; Kanaboshi, H. Cloning and expression of a gene encoding a putative chloroplast w6 fatty acid desaturase of marine Chlamydomonas. Plant Biotechnol. 2000, 17, 167–171. [Google Scholar] [CrossRef] [Green Version]
- Salama, E.-S.; Kim, H.C.; Abou-Shanab, R.A.; Ji, M.K.; Oh, Y.K.; Kim, S.H.; Jeon, B.H. Biomass, lipid content, and fatty acid composition of freshwater Chlamydomonas mexicana and Scenedesmus obliquus grown under salt stress. Bioprocess Biosyst. Eng. 2013, 36, 827–833. [Google Scholar] [CrossRef]
- McManus, H.A.; Lewis, L.A. Molecular Phylogenetic Relationships in the Freshwater Family Hydrodictyaceae (Sphaeropleales, Chlorophyceae), With An Emphasis On Pediastrum Duplex(1). J. Phycol. 2011, 47, 152–163. [Google Scholar] [CrossRef] [PubMed]
- Pröschold, T.; Darienko, T. Choricystis and Lewiniosphaera gen. nov. (Trebouxiophyceae Chlorophyta), two different green algal endosymbionts in freshwater sponges. Symbiosis 2020, 82, 175–188. [Google Scholar] [CrossRef] [PubMed]
- Arteaga-Flórez, C.; Fernández-Rodríguez, V.; Londoño-Mesa, M.H. First record of the polychaete Ficopomatus uschakovi (Pillai, 1960) (Annelida, Serpulidae) in the Colombian Caribbean, South America. Zookeys 2014, 371, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simon, C.; Kara, J.; du Toit, A.; van Rensburg, H.; Naidoo, C.; Matthee, C.A. Reeling them in: Taxonomy of marine annelids used as bait by anglers in the Western Cape Province, South Africa. PeerJ 2021, 9, e11847. [Google Scholar] [CrossRef] [PubMed]
- Stekhoven, F.M.S.; van der Velde, G.; Lee, T.H.; Bottrill, A.R. Proteomic study of the brackish water mussel Mytilopsis leucophaeata. Zool Stud. 2015, 54, e22. [Google Scholar] [CrossRef] [Green Version]
- Sousa, R.; Novais, A.; Costa, R.; Strayer, D.L. Invasive bivalves in fresh waters: Impacts from individuals to ecosystems and possible control strategies. Hydrobiologia 2014, 735, 233–251. [Google Scholar] [CrossRef] [Green Version]
- Kraeuter, J.N.; Buckner, S.; Powell, E.N. A note on a spawner-recruit relationship for a heavily exploited bivalve: The case of northern quahogs (hard clams), Mercenaria mercenaria in Great South Bay New York. J. Shellfish Res. 2005, 24, 1043–1052. [Google Scholar]
- Vellend, M. Conceptual synthesis in community ecology. Q Rev. Biol. 2010, 85, 183–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vellend, M.; Srivastava, D.S.; Anderson, K.M.; Brown, C.D.; Jankowski, J.E.; Kleynhans, E.J.; Kraft, N.J.B.; Letaw, A.D.; Macdonald, A.A.M.; Maclean, J.E.; et al. Assessing the relative importance of neutral stochasticity in ecological communities. Oikos 2014, 123, 1420–1430. [Google Scholar] [CrossRef]
- Ofiteru, I.D.; Lunn, M.; Curtis, T.P.; Wells, G.F.; Criddle, C.S.; Francis, C.A.; Sloan, W.T. Combined niche and neutral effects in a microbial wastewater treatment community. Proc. Natl. Acad. Sci. USA 2010, 107, 15345–15350. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Liu, W.; Deng, Y.; Jiang, Y.-H.; Xue, K.; He, Z.; Van Nostrand, J.D.; Wu, L.; Yang, Y.; Wang, A. Stochastic assembly leads to alternative communities with distinct functions in a bioreactor microbial community. mBio 2013, 4, e00584–e00612. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Deng, Y.; Zhang, P.; Xue, K.; Liang, Y.; Van Nostrand, J.D.; Yang, Y.; He, Z.; Wu, L.; Stahl, D.A.; et al. Stochasticity, succession, and environmental perturbations in a fluidic ecosystem. Proc. Natl. Acad. Sci. USA 2014, 111, E836–E845. [Google Scholar] [CrossRef] [Green Version]
- Lozupone, C.A.; Knight, R. Global patterns in bacterial diversity. Proc. Natl. Acad. Sci. USA 2007, 104, 11436–11440. [Google Scholar] [CrossRef] [Green Version]
- Cloern, J.E.; Jassby, A.D.; Schraga, T.S.; Nejad, E.; Martin, C. Ecosystem variability along the estuarine salinity gradient: Examples from long-term study of San Francisco Bay. Limnol. Oceanogr. 2017, 62, 272–291. [Google Scholar] [CrossRef] [Green Version]
- Tee, H.S.; Waite, D.; Lear, G.; Handley, K.M. Microbial river-to-sea continuum: Gradients in benthic and planktonic diversity, osmoregulation and nutrient cycling. Microbiome 2021, 9, 190. [Google Scholar] [CrossRef]
- Chave, J. Neutral theory and community ecology. Ecol. Lett. 2004, 7, 241–253. [Google Scholar] [CrossRef]
- Stegen, J.C.; Lin, X.; Fredrickson, J.K.; Konopka, A.E. Estimating and mapping ecological processes influencing microbial community assembly. Front. Microbiol. 2015, 6, 370. [Google Scholar] [CrossRef] [Green Version]
- Chase, J.M. Stochastic community assembly causes higher biodiversity in more productive environments. Science 2010, 328, 1388–1391. [Google Scholar] [CrossRef] [Green Version]
- Chase, J.M.; Myers, J.A. Disentangling the importance of ecological niches from stochastic processes across scales. Philos. Trans. R Soc. Lond. B Biol. Sci. 2011, 366, 2351–2363. [Google Scholar] [CrossRef] [Green Version]
- Stegen, J.C.; Fredrickson, J.K.; Wilkins, M.J.; Konopka, A.E.; Nelson, W.C.; Arntzen, E.V.; Chrisler, W.B.; Chu, R.K.; Danczak, R.E.; Fansler, S.J.; et al. Groundwater-surface water mixing shifts ecological assembly processes and stimulates organic carbon turnover. Nat. Commun. 2016, 7, 11237. [Google Scholar] [CrossRef] [Green Version]
- Hubbell, S.P. Neutral theory in community ecology and the hypothesis of functional equivalence. Funct. Ecol. 2005, 19, 166–172. [Google Scholar] [CrossRef]
- Gravel, D.; Canham, C.D.; Beaudet, M.; Messier, C. Reconciling niche and neutrality: The continuum hypothesis. Ecol. Lett. 2006, 9, 399–409. [Google Scholar] [CrossRef] [Green Version]
- Gewin, V. Beyond neutrality-ecology finds its niche. PLoS Biol. 2006, 4, e278. [Google Scholar] [CrossRef] [Green Version]
- Adler, P.B.; HilleRisLambers, J.; Levine, J.M. A niche for neutrality. Ecol. Lett. 2007, 10, 95–104. [Google Scholar] [CrossRef] [Green Version]
- Matthews, T.J.; Whittaker, R.J. Neutral theory and the species abundance distribution: Recent developments and prospects for unifying niche and neutral perspectives. Ecol. Evol. 2014, 4, 2263–2277. [Google Scholar] [CrossRef]
- Rosindell, J.; Hubbell, S.P.; He, F.; Harmon, L.J.; Etienne, R.S. The case for ecological neutral theory. Trends Ecol. Evol. 2012, 27, 203–208. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample Name | Shannon Index | Inv_Simpson Index | Observed Richness | Chao1 Index | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| O | A | R | C | O | A | R | C | O | A | R | C | O | A | R | C | |
| Sanya_1_3 um | 5.63 | 3.89 | 5.98 | 5.22 | 121.31 | 36.31 | 315.30 | 153.60 | 1008 | 107 | 606 | 295 | 1136.69 | 108.67 | 719.05 | 313.45 |
| Sanya_1_0.22 um | 5.20 | 3.75 | 5.72 | 5.02 | 68.07 | 26.98 | 229.66 | 123.64 | 838 | 103 | 487 | 248 | 966.18 | 114.25 | 581.94 | 265.77 |
| Sanya_2_3 um | 4.91 | 3.87 | 5.51 | 4.79 | 59.99 | 32.72 | 172.11 | 94.12 | 827 | 130 | 451 | 246 | 1085.62 | 141.38 | 647.87 | 293.04 |
| Sanya_2_0.22 um | 4.69 | 3.86 | 5.48 | 4.75 | 48.65 | 31.47 | 163.53 | 84.95 | 760 | 128 | 404 | 228 | 945.01 | 136.25 | 555.54 | 251.62 |
| Sanya_3_3 um | 4.95 | 3.66 | 6.01 | 4.87 | 40.77 | 21.05 | 311.77 | 88.92 | 1073 | 147 | 640 | 286 | 1247.25 | 154.80 | 766.91 | 322.75 |
| Sanya_3_0.22 um | 4.66 | 3.47 | 5.94 | 4.98 | 26.12 | 14.80 | 278.13 | 107.52 | 1030 | 155 | 598 | 277 | 1232.58 | 176.00 | 736.60 | 315.33 |
| Sanya_4_3 um | 4.63 | 3.70 | 5.71 | 4.64 | 39.46 | 24.45 | 196.82 | 69.31 | 920 | 132 | 525 | 263 | 1177.01 | 135.21 | 714.07 | 335.33 |
| Sanya_4_0.22 um | 4.23 | 3.45 | 5.53 | 4.63 | 21.01 | 14.73 | 160.12 | 68.25 | 828 | 141 | 443 | 244 | 1130.58 | 156.30 | 727.29 | 271.03 |
| Sanya_5_3 um | 4.88 | 3.56 | 5.97 | 4.79 | 42.49 | 20.98 | 292.49 | 94.36 | 969 | 114 | 634 | 221 | 1176.01 | 124.11 | 799.00 | 248.19 |
| Sanya_5_0.22 um | 4.76 | 3.54 | 5.74 | 4.74 | 40.08 | 20.62 | 223.25 | 91.03 | 897 | 125 | 546 | 226 | 1242.24 | 153.11 | 774.30 | 312.25 |
| Sanya_6_3 um | 3.99 | 2.72 | 5.69 | 4.52 | 14.85 | 8.56 | 218.60 | 70.84 | 719 | 79 | 474 | 166 | 850.00 | 88.00 | 583.00 | 175.10 |
| Sanya_6_0.22 um | 4.16 | 2.80 | 5.77 | 4.69 | 12.95 | 7.09 | 243.78 | 86.60 | 758 | 85 | 494 | 179 | 891.47 | 90.60 | 606.50 | 191.75 |
| Sample Name | Shannon Index | Inv_Simpson Index | Observed Richness | Chao1 Index | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| O | A | R | C | O | A | R | C | O | A | R | C | O | A | R | C | |
| Sanya_1_3 um | 4.92 | 3.59 | 5.41 | 4.81 | 58.19 | 25.42 | 178.79 | 103.12 | 608 | 84 | 337 | 187 | 689.43 | 86.63 | 396.44 | 206.43 |
| Sanya_1_0.22 um | 3.32 | 2.29 | 5.05 | 4.42 | 5.46 | 3.60 | 121.42 | 62.44 | 484 | 78 | 247 | 159 | 583.00 | 94.50 | 335.14 | 165.32 |
| Sanya_2_3 um | 4.26 | 3.48 | 4.95 | 4.46 | 29.80 | 19.88 | 99.85 | 67.56 | 526 | 115 | 232 | 179 | 657.21 | 130.55 | 290.72 | 238.13 |
| Sanya_2_0.22 um | 3.94 | 3.26 | 4.92 | 4.46 | 19.22 | 14.18 | 95.77 | 62.23 | 509 | 119 | 226 | 164 | 612.89 | 122.60 | 305.03 | 185.94 |
| Sanya_3_3 um | 3.28 | 2.64 | 4.75 | 4.05 | 8.92 | 6.92 | 83.51 | 40.97 | 439 | 116 | 184 | 139 | 535.30 | 134.40 | 227.56 | 168.75 |
| Sanya_3_0.22 um | 3.41 | 2.81 | 4.80 | 4.05 | 10.94 | 8.55 | 91.37 | 36.86 | 464 | 121 | 193 | 150 | 581.00 | 123.65 | 269.61 | 203.81 |
| Sanya_4_3 um | 3.64 | 2.96 | 4.49 | 4.20 | 11.38 | 8.29 | 61.16 | 51.72 | 389 | 95 | 163 | 131 | 541.63 | 137.17 | 256.26 | 148.77 |
| Sanya_4_0.22 um | 3.23 | 2.63 | 4.56 | 4.18 | 6.31 | 4.95 | 62.28 | 48.30 | 396 | 90 | 171 | 135 | 560.36 | 105.17 | 261.23 | 184.58 |
| Sanya_5_3 um | 4.72 | 3.70 | 5.33 | 4.61 | 44.91 | 25.02 | 156.74 | 77.22 | 648 | 130 | 330 | 188 | 749.97 | 151.08 | 384.60 | 213.09 |
| Sanya_5_0.22 um | 4.40 | 3.32 | 5.28 | 4.55 | 21.59 | 12.42 | 159.36 | 76.25 | 587 | 119 | 300 | 168 | 759.22 | 128.10 | 408.37 | 224.10 |
| Sanya_6_3 um | 3.68 | 2.64 | 5.08 | 4.26 | 10.36 | 6.44 | 127.78 | 58.12 | 453 | 78 | 250 | 125 | 568.50 | 93.17 | 331.36 | 138.60 |
| Sanya_6_0.22 um | 2.06 | 1.49 | 4.62 | 3.90 | 2.58 | 2.16 | 73.16 | 35.75 | 329 | 66 | 161 | 102 | 426.23 | 106.00 | 210.34 | 120.07 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bai, S.; Zhang, J.; Qi, X.; Zeng, J.; Wu, S.; Peng, X. Changes of In Situ Prokaryotic and Eukaryotic Communities in the Upper Sanya River to the Sea over a Nine-Hour Period. Microorganisms 2023, 11, 536. https://doi.org/10.3390/microorganisms11020536
Bai S, Zhang J, Qi X, Zeng J, Wu S, Peng X. Changes of In Situ Prokaryotic and Eukaryotic Communities in the Upper Sanya River to the Sea over a Nine-Hour Period. Microorganisms. 2023; 11(2):536. https://doi.org/10.3390/microorganisms11020536
Chicago/Turabian StyleBai, Shijie, Jian Zhang, Xiaoxue Qi, Juntao Zeng, Shijun Wu, and Xiaotong Peng. 2023. "Changes of In Situ Prokaryotic and Eukaryotic Communities in the Upper Sanya River to the Sea over a Nine-Hour Period" Microorganisms 11, no. 2: 536. https://doi.org/10.3390/microorganisms11020536