
Gut Microbiota and Metabolome Changes in Three Pulmonary Hypertension Rat Models
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals and Reagents
2.2. Establishment of Animal Models
2.3. 16S rRNA Gene Amplification and Multiparallel Sequencing
2.4. Metabolite Extraction
2.5. Metabolome Analysis
2.6. Hematoxylin and Eosin Staining
2.7. Data Analysis
2.8. Statistical Analysis
3. Results
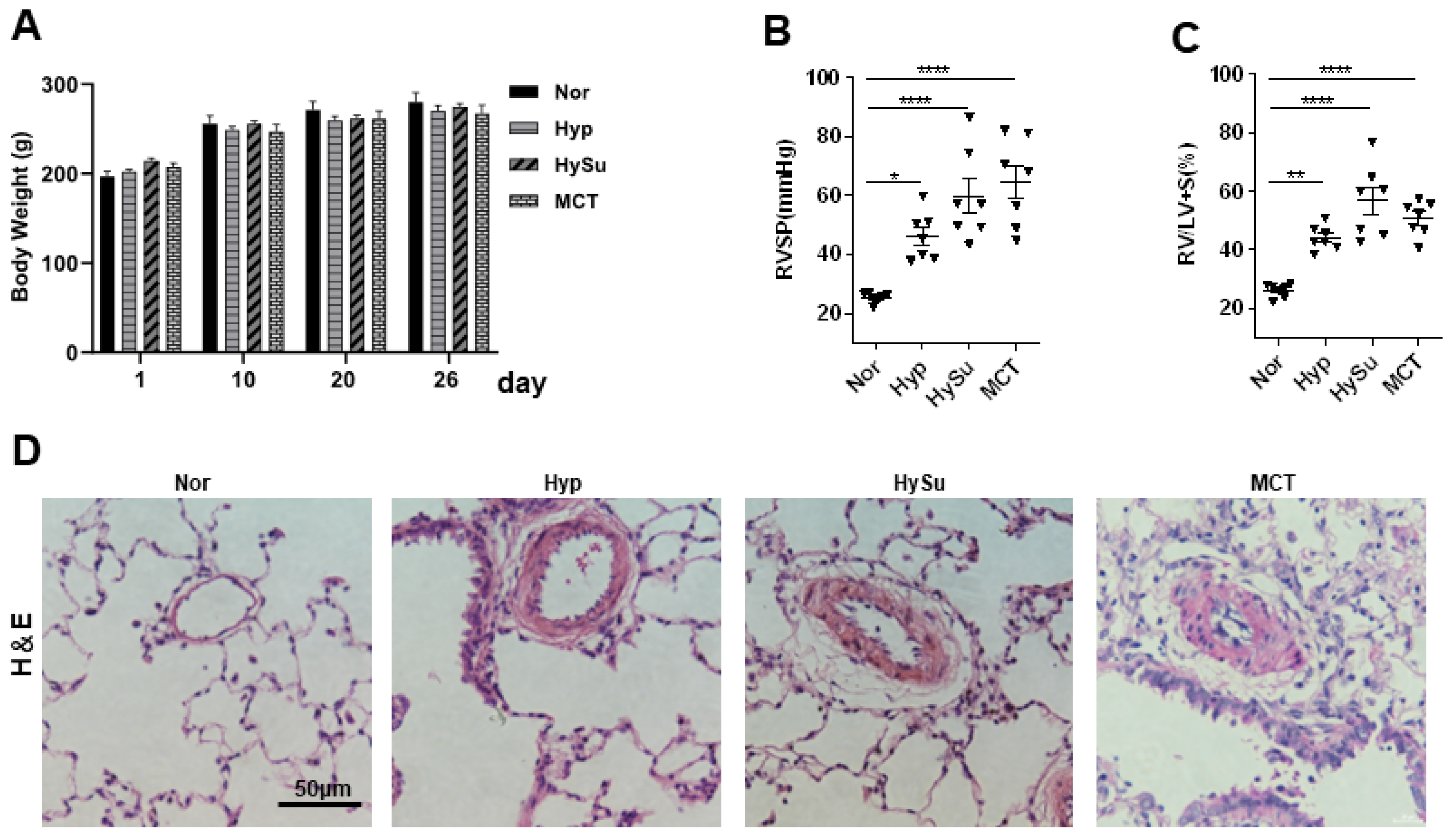
3.1. Animal Profiles
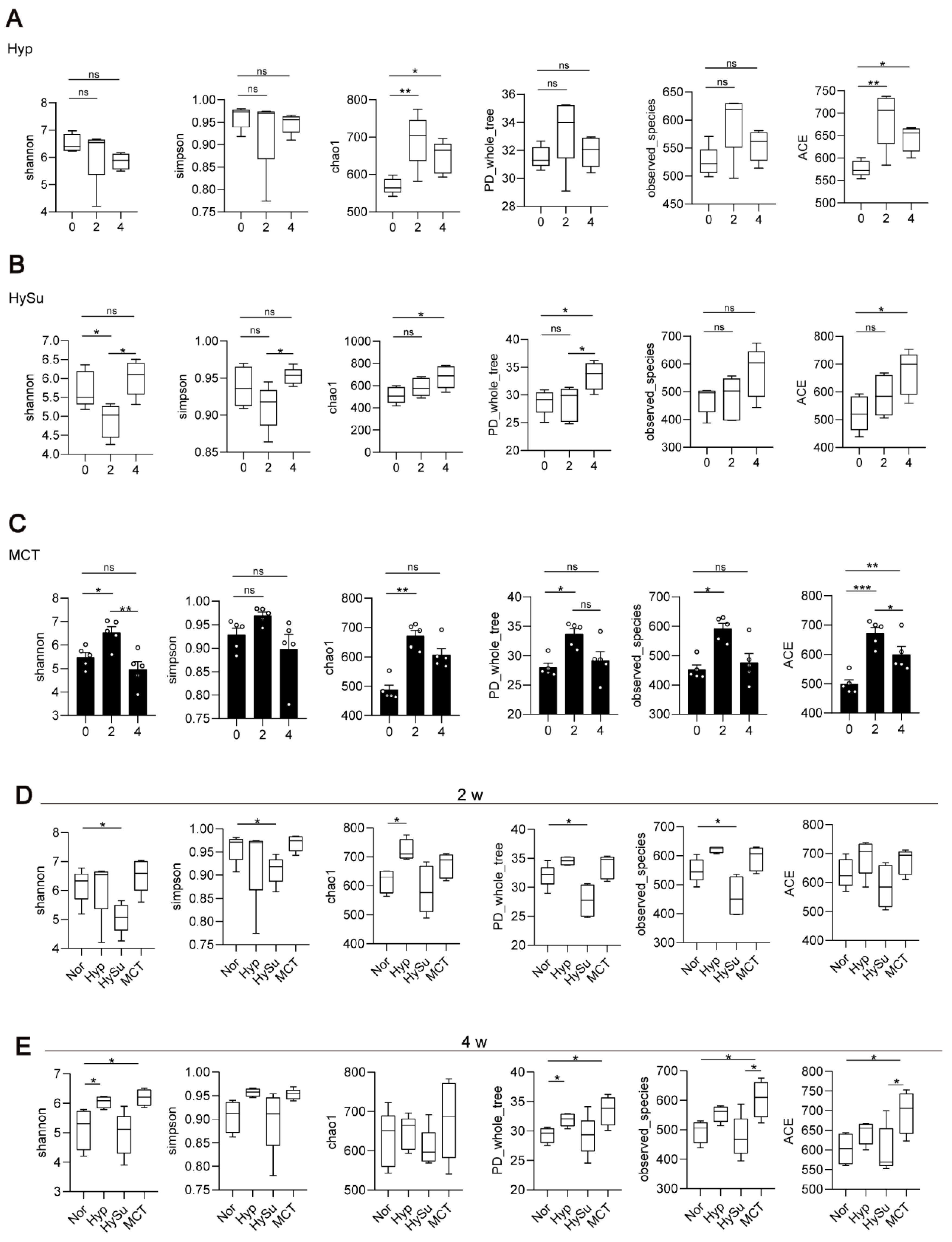
3.2. Overall Microbial Content of Nor, Hyp, HySu, and MCT Rats
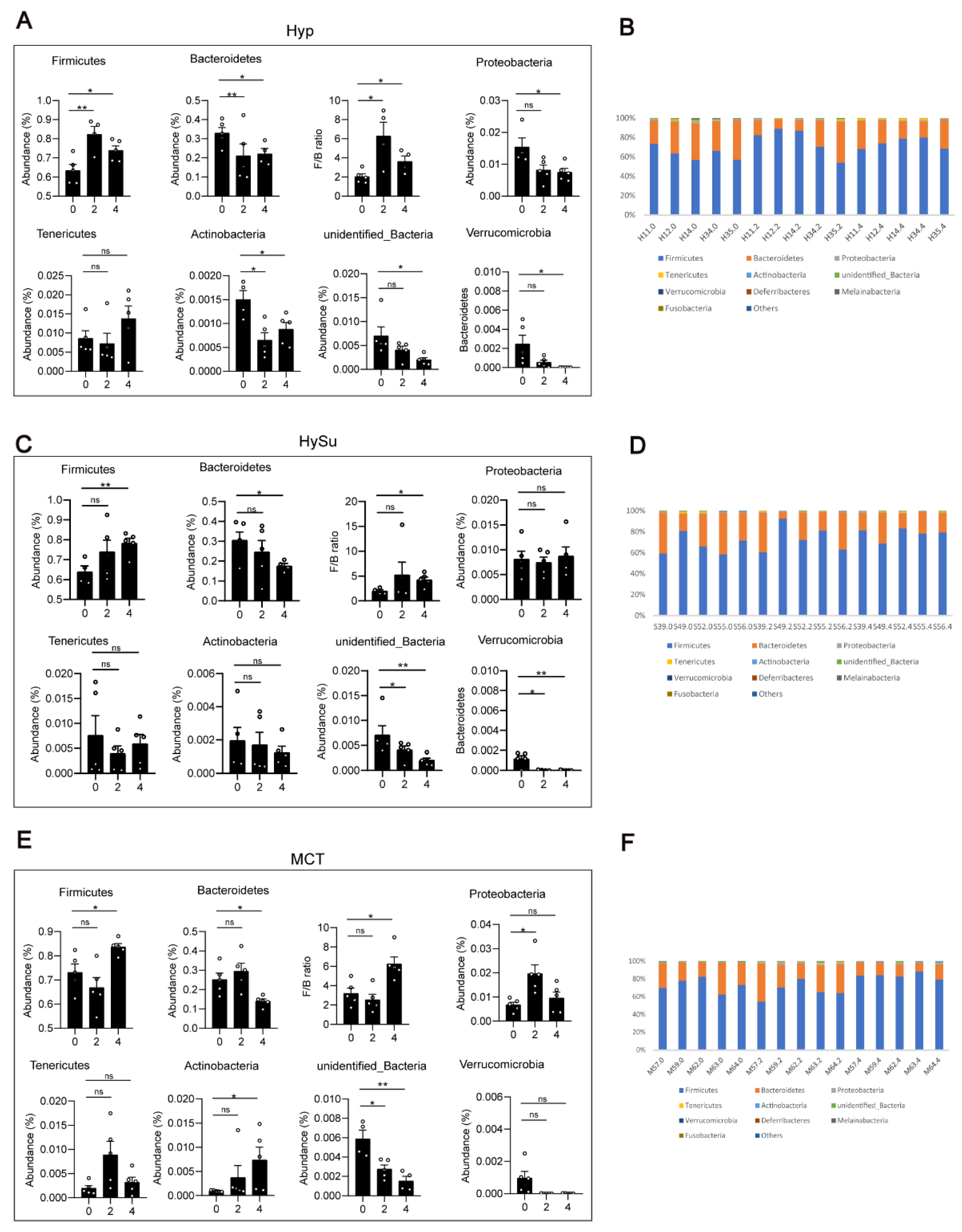
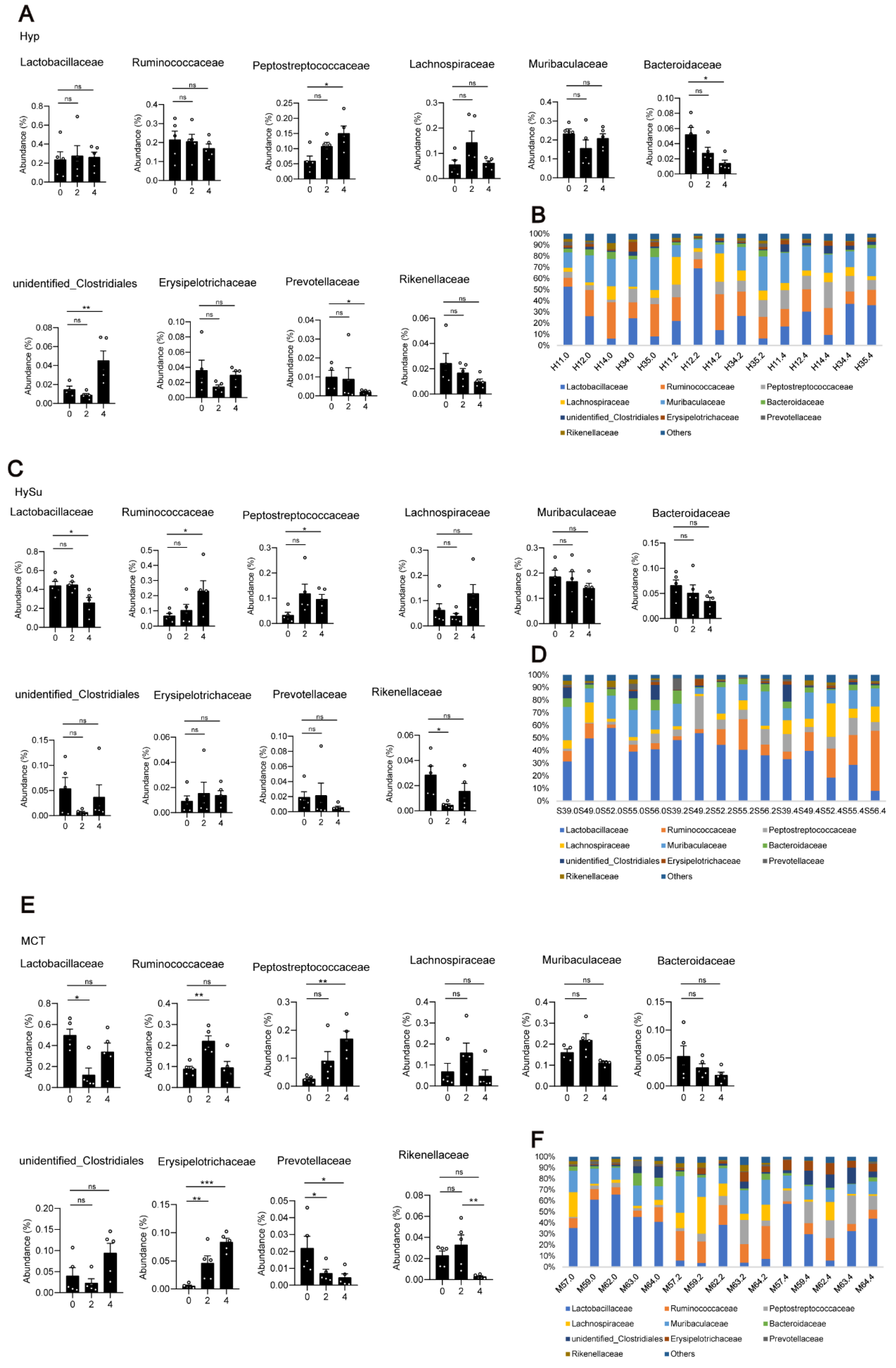
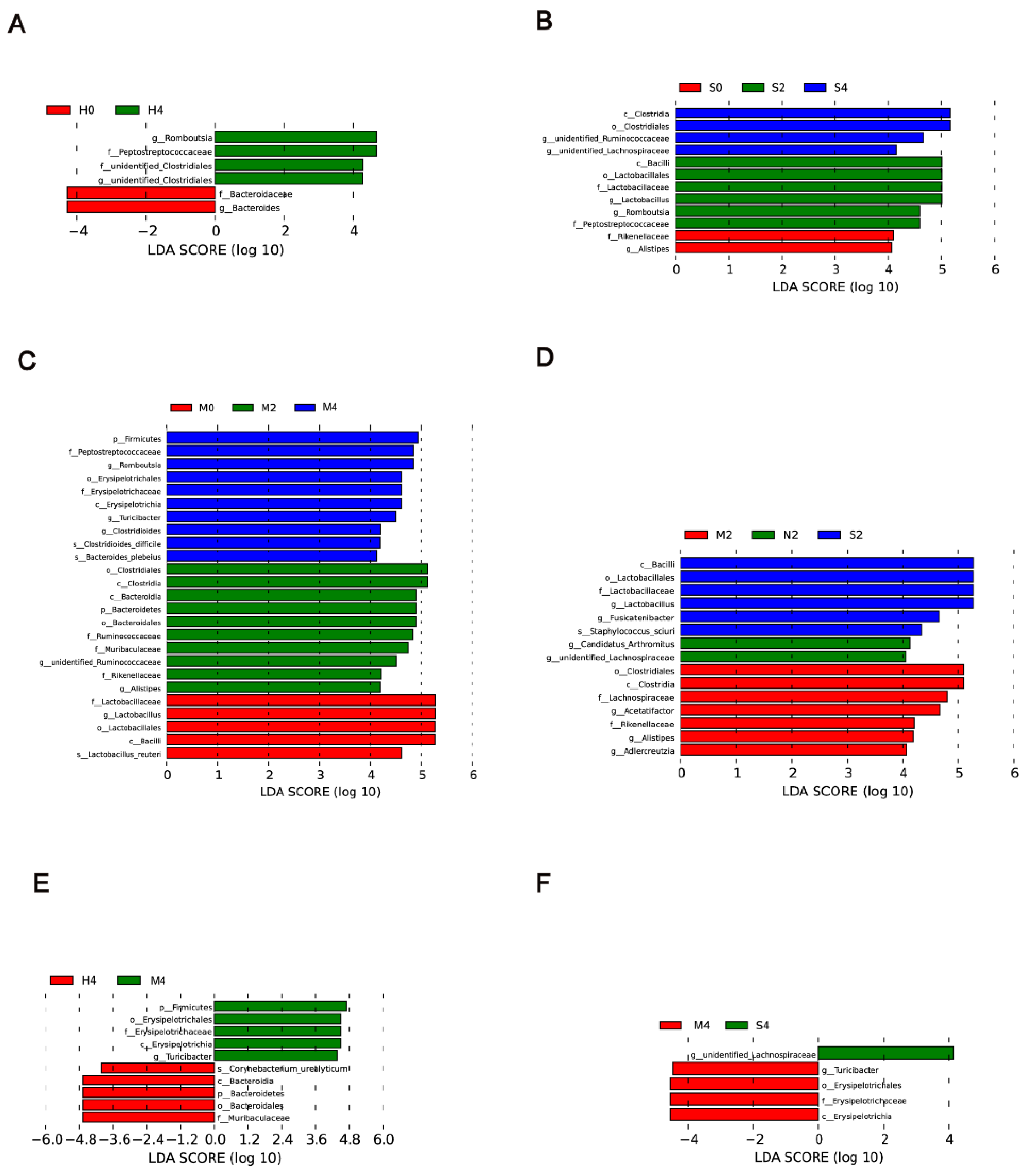
3.3. Alterations in the Gut Microbiota in Response to Hypoxia, Hypoxia/Sugen 5416, and MCT
3.4. Potential Functions of the Gut Microbiota in Hypoxia-Induced, Hypoxia/Sugen 5416-Induced, and MCT-Induced Rats
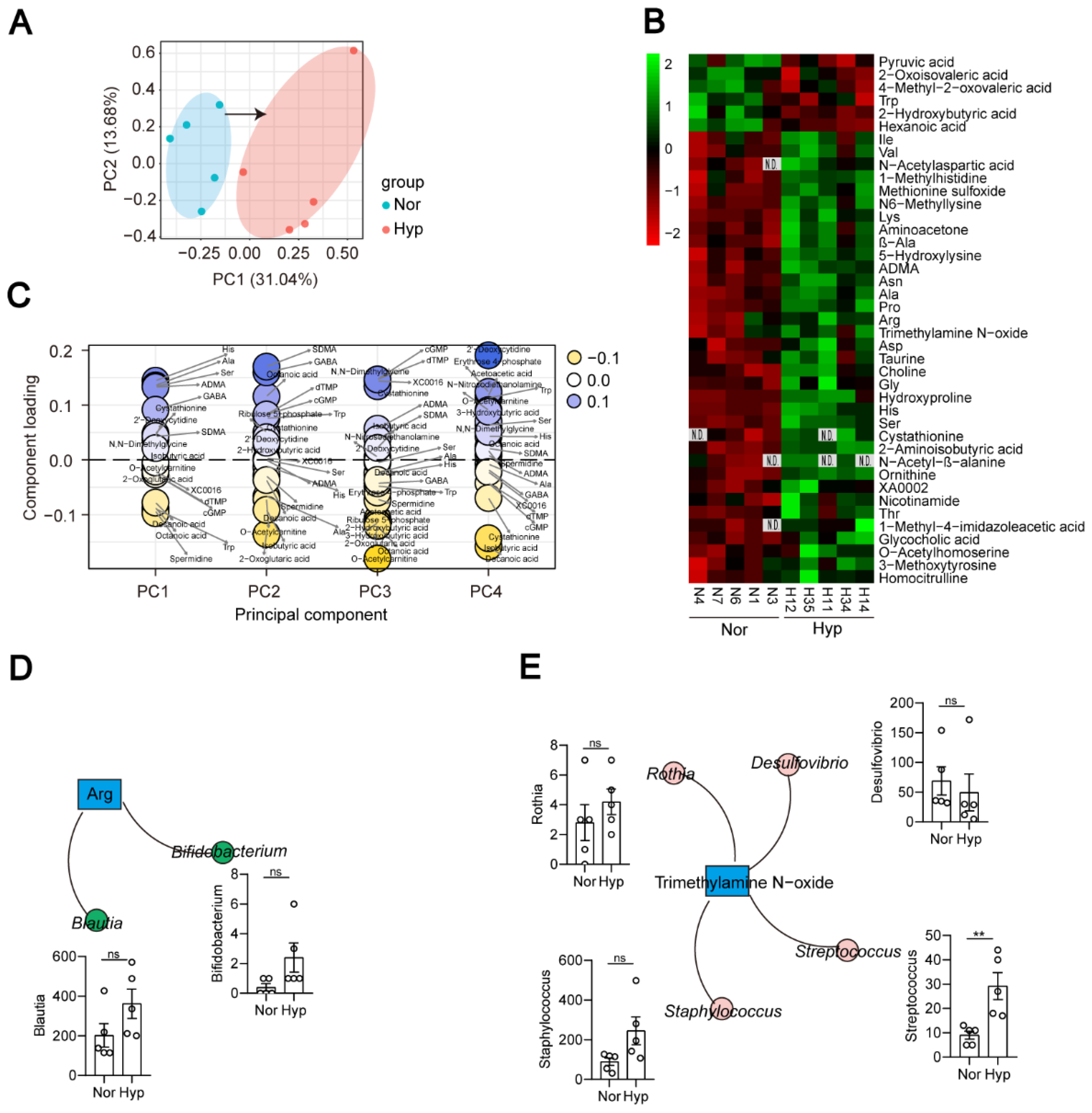
3.5. Functional Indicators of the Hypoxia-Induced PH Rat Plasma Metabolome
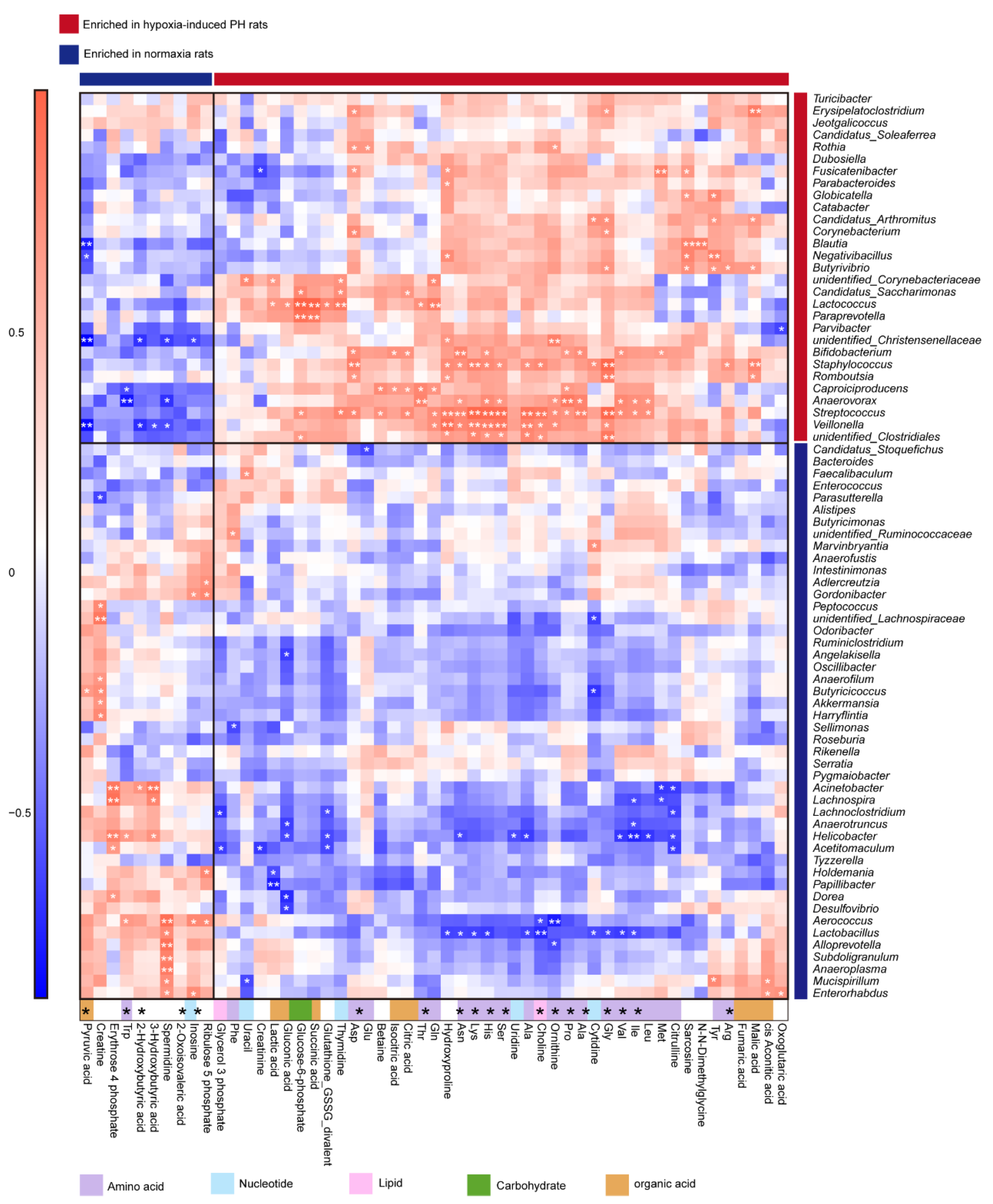
3.6. Correlation Analysis of Microbial Diversity with Plasma Metabolites in Hypoxia-Induced PH Rats
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Pulido, T.; Adzerikho, I.; Channick, R.N.; Delcroix, M.; Galiè, N.; Ghofrani, A.; Jansa, P.; Jing, Z.-C.; Le Brun, F.-O.; Mehta, S.; et al. Macitentan and morbidity and mortality in pulmonary arterial hypertension. N. Engl. J. Med. 2013, 369, 809–818. [Google Scholar] [CrossRef]
- Del Pozo, R.; Hernandez Gonzalez, I.; Escribano-Subias, P. The prostacyclin pathway in pulmonary arterial hypertension: A clinical review. Expert Rev. Respir. Med. 2017, 11, 491–503. [Google Scholar] [CrossRef]
- Liu, C.; Chen, J.; Gao, Y.; Deng, B.; Liu, K. Endothelin receptor antagonists for pulmonary arterial hypertension. Cochrane Database Syst. Rev. 2009, 1, CD004434. [Google Scholar] [CrossRef]
- Barnes, H.; Brown, Z.; Burns, A.; Williams, T. Phosphodiesterase 5 inhibitors for pulmonary hypertension. Cochrane Database Syst. Rev. 2019, 1, CD012621. [Google Scholar] [CrossRef]
- Beghetti, M.; Gorenflo, M.; Ivy, D.D.; Moledina, S.; Bonnet, D. Treatment of pediatric pulmonary arterial hypertension: A focus on the NO-sGC-cGMP pathway. Pediatr. Pulmonol. 2019, 54, 1516–1526. [Google Scholar] [CrossRef]
- Lozupone, C.A.; Stombaugh, J.I.; Gordon, J.I.; Jansson, J.K.; Knight, R. Diversity, stability and resilience of the human gut microbiota. Nature 2012, 489, 220–230. [Google Scholar] [CrossRef]
- Wang, Z.; Zhao, Y. Gut microbiota derived metabolites in cardiovascular health and disease. Protein Cell 2018, 9, 416–431. [Google Scholar] [CrossRef]
- Wong, S.H.; Yu, J. Gut microbiota in colorectal cancer: Mechanisms of action and clinical applications. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 690–704. [Google Scholar] [CrossRef]
- Nishida, A.; Inoue, R.; Inatomi, O.; Bamba, S.; Naito, Y.; Andoh, A. Gut microbiota in the pathogenesis of inflammatory bowel disease. Clin. J. Gastroenterol. 2018, 11, 1–10. [Google Scholar] [CrossRef]
- Bhattarai, Y.; Muniz Pedrogo, D.A.; Kashyap, P.C. Irritable bowel syndrome: A gut microbiota-related disorder? Am. J. Physiol. Gastrointest. Liver Physiol. 2017, 312, G52–G62. [Google Scholar] [CrossRef]
- Bolla, A.M.; Caretto, A.; Laurenzi, A.; Scavini, M.; Piemonti, L. Low-Carb and Ketogenic Diets in Type 1 and Type 2 Diabetes. Nutrients 2019, 11, 962. [Google Scholar] [CrossRef]
- Horta-Baas, G.; Romero-Figueroa, M.D.S.; Montiel-Jarquín, A.J.; Pizano-Zárate, M.L.; García-Mena, J.; Ramírez-Durán, N. Intestinal Dysbiosis and Rheumatoid Arthritis: A Link between Gut Microbiota and the Pathogenesis of Rheumatoid Arthritis. J. Immunol. Res. 2017, 2017, 4835189. [Google Scholar] [CrossRef]
- Kim, S.; Rigatto, K.; Gazzana, M.B.; Knorst, M.M.; Richards, E.M.; Pepine, C.J.; Raizada, M.K. Altered Gut Microbiome Profile in Patients With Pulmonary Arterial Hypertension. Hypertension 2020, 75, 1063–1071. [Google Scholar] [CrossRef]
- Bowerman, K.L.; Rehman, S.F.; Vaughan, A.; Lachner, N.; Budden, K.F.; Kim, R.Y.; Wood, D.L.A.; Gellatly, S.L.; Shukla, S.D.; Wood, L.G.; et al. Disease-associated gut microbiome and metabolome changes in patients with chronic obstructive pulmonary disease. Nat. Commun. 2020, 11, 5886. [Google Scholar] [CrossRef]
- Sharma, R.K.; Oliveira, A.C.; Yang, T.; Kim, S.; Zubcevic, J.; Aquino, V.; Lobaton, G.O.; Goel, R.; Richards, E.M.; Raizada, M.K. Pulmonary arterial hypertension-associated changes in gut pathology and microbiota. ERJ Open Res. 2020, 6, 00253-2019. [Google Scholar] [CrossRef]
- Callejo, M.; Mondejar-Parreño, G.; Barreira, B.; Izquierdo-Garcia, J.L.; Morales-Cano, D.; Esquivel-Ruiz, S.; Moreno, L.; Cogolludo, Á.; Duarte, J.; Perez-Vizcaino, F. Pulmonary Arterial Hypertension Affects the Rat Gut Microbiome. Sci. Rep. 2018, 8, 9681. [Google Scholar] [CrossRef]
- Rutten, E.P.A.; Lenaerts, K.; Buurman, W.A.; Wouters, E.F.M. Disturbed intestinal integrity in patients with COPD: Effects of activities of daily living. Chest 2014, 145, 245–252. [Google Scholar] [CrossRef]
- Keely, S.; Talley, N.J.; Hansbro, P.M. Pulmonary-intestinal cross-talk in mucosal inflammatory disease. Mucosal. Immunol. 2012, 5, 7–18. [Google Scholar] [CrossRef]
- Segal, L.N.; Clemente, J.C.; Tsay, J.-C.J.; Koralov, S.B.; Keller, B.C.; Wu, B.G.; Alison, M.; Shen, N.; Ghedin, E.; Morris, A.; et al. Enrichment of the lung microbiome with oral taxa is associated with lung inflammation of a Th17 phenotype. Nat. Microbiol. 2016, 1, 16031. [Google Scholar] [CrossRef]
- Schiavi, E.; Plattner, S.; Rodriguez-Perez, N.; Barcik, W.; Frei, R.; Ferstl, R.; Kurnik-Lucka, M.; Groeger, D.; Grant, R.; Roper, J.; et al. Exopolysaccharide from Bifidobacterium longum subsp. longum 35624 modulates murine allergic airway responses. Benef. Microbes 2018, 9, 761–773. [Google Scholar] [CrossRef]
- Vierbuchen, T.; Bang, C.; Rosigkeit, H.; Schmitz, R.A.; Heine, H. The Human-Associated Archaeon Methanosphaera stadtmanae Is Recognized through Its RNA and Induces TLR8-Dependent NLRP3 Inflammasome Activation. Front. Immunol. 2017, 8, 1535. [Google Scholar] [CrossRef]
- Freer, G.; Maggi, F.; Pifferi, M.; Di Cicco, M.; Peroni, D.G.; Pistello, M. The Virome and Its Major Component, Anellovirus, a Convoluted System Molding Human Immune Defenses and Possibly Affecting the Development of Asthma and Respiratory Diseases in Childhood. Front. Microbiol. 2018, 9, 686. [Google Scholar] [CrossRef]
- Budden, K.F.; Shukla, S.D.; Rehman, S.F.; Bowerman, K.L.; Keely, S.; Hugenholtz, P.; Armstrong-James, D.P.H.; Adcock, I.M.; Chotirmall, S.H.; Chung, K.F.; et al. Functional effects of the microbiota in chronic respiratory disease. Lancet Respir. Med. 2019, 7, 907–920. [Google Scholar] [CrossRef]
- Marques, F.Z.; Nelson, E.; Chu, P.-Y.; Horlock, D.; Fiedler, A.; Ziemann, M.; Tan, J.K.; Kuruppu, S.; Rajapakse, N.W.; El-Osta, A.; et al. High-Fiber Diet and Acetate Supplementation Change the Gut Microbiota and Prevent the Development of Hypertension and Heart Failure in Hypertensive Mice. Circulation 2017, 135, 964–977. [Google Scholar] [CrossRef]
- Segal, L.N.; Clemente, J.C.; Wu, B.G.; Wikoff, W.R.; Gao, Z.; Li, Y.; Ko, J.P.; Rom, W.N.; Blaser, M.J.; Weiden, M.D. Randomised, double-blind, placebo-controlled trial with azithromycin selects for anti-inflammatory microbial metabolites in the emphysematous lung. Thorax 2017, 72, 13–22. [Google Scholar] [CrossRef]
- Sanada, T.J.; Hosomi, K.; Shoji, H.; Park, J.; Naito, A.; Ikubo, Y.; Yanagisawa, A.; Kobayashi, T.; Miwa, H.; Suda, R.; et al. Gut microbiota modification suppresses the development of pulmonary arterial hypertension in an SU5416/hypoxia rat model. Pulm. Circ. 2020, 10, 2045894020929147. [Google Scholar] [CrossRef]
- Ooga, T.; Sato, H.; Nagashima, A.; Sasaki, K.; Tomita, M.; Soga, T.; Ohashi, Y. Metabolomic anatomy of an animal model revealing homeostatic imbalances in dyslipidaemia. Mol. Biosyst. 2011, 7, 1217–1223. [Google Scholar] [CrossRef]
- Sugimoto, M.; Hirayama, A.; Robert, M.; Abe, S.; Soga, T.; Tomita, M. Prediction of metabolite identity from accurate mass, migration time prediction and isotopic pattern information in CE-TOFMS data. Electrophoresis 2010, 31, 2311–2318. [Google Scholar] [CrossRef]
- Cardiff, R.D.; Miller, C.H.; Munn, R.J. Manual hematoxylin and eosin staining of mouse tissue sections. Cold Spring Harb. Protoc. 2014, 2014, 655–658. [Google Scholar] [CrossRef]
- Liu, H.X.; Rocha, C.S.; Dandekar, S.; Wan, Y.J. Functional analysis of the relationship between intestinal microbiota and the expression of hepatic genes and pathways during the course of liver regeneration. J. Hepatol. 2016, 64, 641–650. [Google Scholar] [CrossRef]
- Ikubo, Y.; Sanada, T.J.; Hosomi, K.; Park, J.; Naito, A.; Shoji, H.; Misawa, T.; Suda, R.; Sekine, A.; Sugiura, T.; et al. Altered gut microbiota and its association with inflammation in patients with chronic thromboembolic pulmonary hypertension: A single-center observational study in Japan. BMC Pulm. Med. 2022, 22, 138. [Google Scholar] [CrossRef]
- Hong, W.; Mo, Q.; Wang, L.; Peng, F.; Zhou, Y.; Zou, W.; Sun, R.; Liang, C.; Zheng, M.; Li, H.; et al. Changes in the gut microbiome and metabolome in a rat model of pulmonary arterial hypertension. Bioengineered 2021, 12, 5173–5183. [Google Scholar] [CrossRef]
- Kaakoush, N.O. Insights into the Role of Erysipelotrichaceae in the Human Host. Front. Cell. Infect. Microbiol. 2015, 5, 84. [Google Scholar] [CrossRef]
- Dinh, D.M.; Volpe, G.E.; Duffalo, C.; Bhalchandra, S.; Tai, A.K.; Kane, A.V.; Wanke, C.A.; Ward, H.D. Intestinal microbiota, microbial translocation, and systemic inflammation in chronic HIV infection. J. Infect. Dis. 2015, 211, 19–27. [Google Scholar] [CrossRef]
- Mohammed, J.P.; Mattner, J. Autoimmune disease triggered by infection with alphaproteobacteria. Expert Rev. Clin. Immunol. 2009, 5, 369–379. [Google Scholar] [CrossRef]
- Luo, L.; Chen, Q.; Yang, L.; Zhang, Z.; Xu, J.; Gou, D. MSCs Therapy Reverse the Gut Microbiota in Hypoxia-Induced Pulmonary Hypertension Mice. Front. Physiol. 2021, 12, 712139. [Google Scholar] [CrossRef]
- Kim, C.H.; Park, J.; Kim, M. Gut microbiota-derived short-chain Fatty acids, T cells, and inflammation. Immune Netw. 2014, 14, 277–288. [Google Scholar] [CrossRef]
- Wang, L.; Liao, Y.; Yang, R.; Zhu, Z.; Zhang, L.; Wu, Z.; Sun, X. An engineered probiotic secreting Sj16 ameliorates colitis via Ruminococcaceae/butyrate/retinoic acid axis. Bioeng. Transl. Med. 2021, 6, e10219. [Google Scholar] [CrossRef]
- Zou, F.; Qiu, Y.; Huang, Y.; Zou, H.; Cheng, X.; Niu, Q.; Luo, A.; Sun, J. Effects of short-chain fatty acids in inhibiting HDAC and activating p38 MAPK are critical for promoting B10 cell generation and function. Cell Death Dis. 2021, 12, 582. [Google Scholar] [CrossRef]
- Guo, M.; Zhang, M.; Cao, X.; Fang, X.; Li, K.; Qin, L.; He, Y.; Zhao, J.; Xu, Y.; Liu, X.; et al. Notch4 mediates vascular remodeling via ERK/JNK/P38 MAPK signaling pathways in hypoxic pulmonary hypertension. Respir. Res. 2022, 23, 6. [Google Scholar] [CrossRef]
- Ouyang, S.; Chen, W.; Gaofeng, Z.; Changcheng, L.; Guoping, T.; Minyan, Z.; Yang, L.; Min, Y.; Luo, J. Cyanidin3Obetaglucoside protects against pulmonary artery hypertension induced by monocrotaline via the TGFbeta1/p38 MAPK/CREB signaling pathway. Mol. Med. Rep. 2021, 23, 1–12. [Google Scholar] [CrossRef]
- Vanderpool, R.R.; Tang, H.; Rischard, F.; Yuan, J.X. Is p38 MAPK a Dark Force in Right Ventricular Hypertrophy and Failure in Pulmonary Arterial Hypertension? Am. J. Respir. Cell Mol. Biol. 2017, 57, 506–508. [Google Scholar] [CrossRef]
- Aktories, K.; Schwan, C.; Jank, T. Clostridium difficile Toxin Biology. Annu. Rev. Microbiol. 2017, 71, 281–307. [Google Scholar] [CrossRef]
- Rhodes, C.; Ghataorhe, P.; Wharton, J.; Rue-Albrecht, K.; Hadinnapola, C.; Watson, G.; Bleda, M.; Haimel, M.; Coghlan, G.; Corris, P.A.; et al. Plasma Metabolomics Implicates Modified Transfer RNAs and Altered Bioenergetics in the Outcomes of Pulmonary Arterial Hypertension. Circulation 2017, 135, 460–475. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Luo, L.; Yin, H.; Gou, D. Gut Microbiota and Metabolome Changes in Three Pulmonary Hypertension Rat Models. Microorganisms 2023, 11, 472. https://doi.org/10.3390/microorganisms11020472
Luo L, Yin H, Gou D. Gut Microbiota and Metabolome Changes in Three Pulmonary Hypertension Rat Models. Microorganisms. 2023; 11(2):472. https://doi.org/10.3390/microorganisms11020472
Chicago/Turabian StyleLuo, Lingjie, Haoyang Yin, and Deming Gou. 2023. "Gut Microbiota and Metabolome Changes in Three Pulmonary Hypertension Rat Models" Microorganisms 11, no. 2: 472. https://doi.org/10.3390/microorganisms11020472