


Inferring Bacterial Community Interactions and Functionalities Associated with Osteopenia and Osteoporosis in Taiwanese Postmenopausal Women

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Characterization of Healthy Participants and Patients
2.2. Sample Collection and Genomic DNA Extraction
2.3. Sequencing, Library Construction, and Microbial Community Analysis
2.4. Functional Profiling Analysis among the Experimental Groups Based on 16S rRNA Gene Signatures
3. Results
3.1. Sequencing and ASV Analysis among the Experimental Groups (Normal Control, Osteopenia, and Osteoporosis) Based on 16S rRNA Gene Signatures
3.2. Bacterial Diversity Analysis among the Experimental Groups Based on 16S rRNA Gene Signatures
3.3. Bacterial Community Profiling and Pattern Analysis at Taxonomic Levels in the Experimental Groups Based on 16S rRNA Gene Signatures
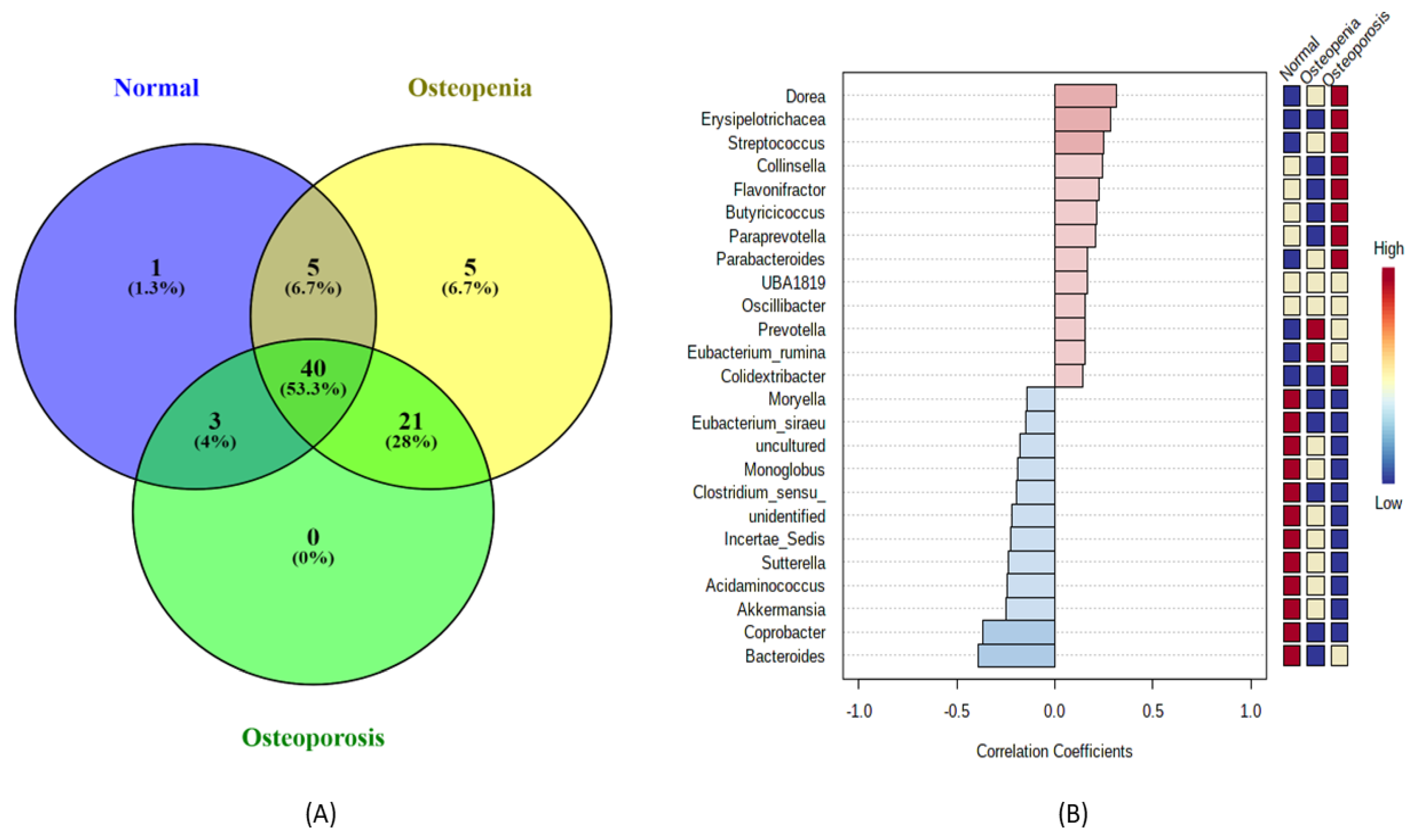
3.4. Bacterial Community Shift Analysis at the Genus Level among the Experimental Groups
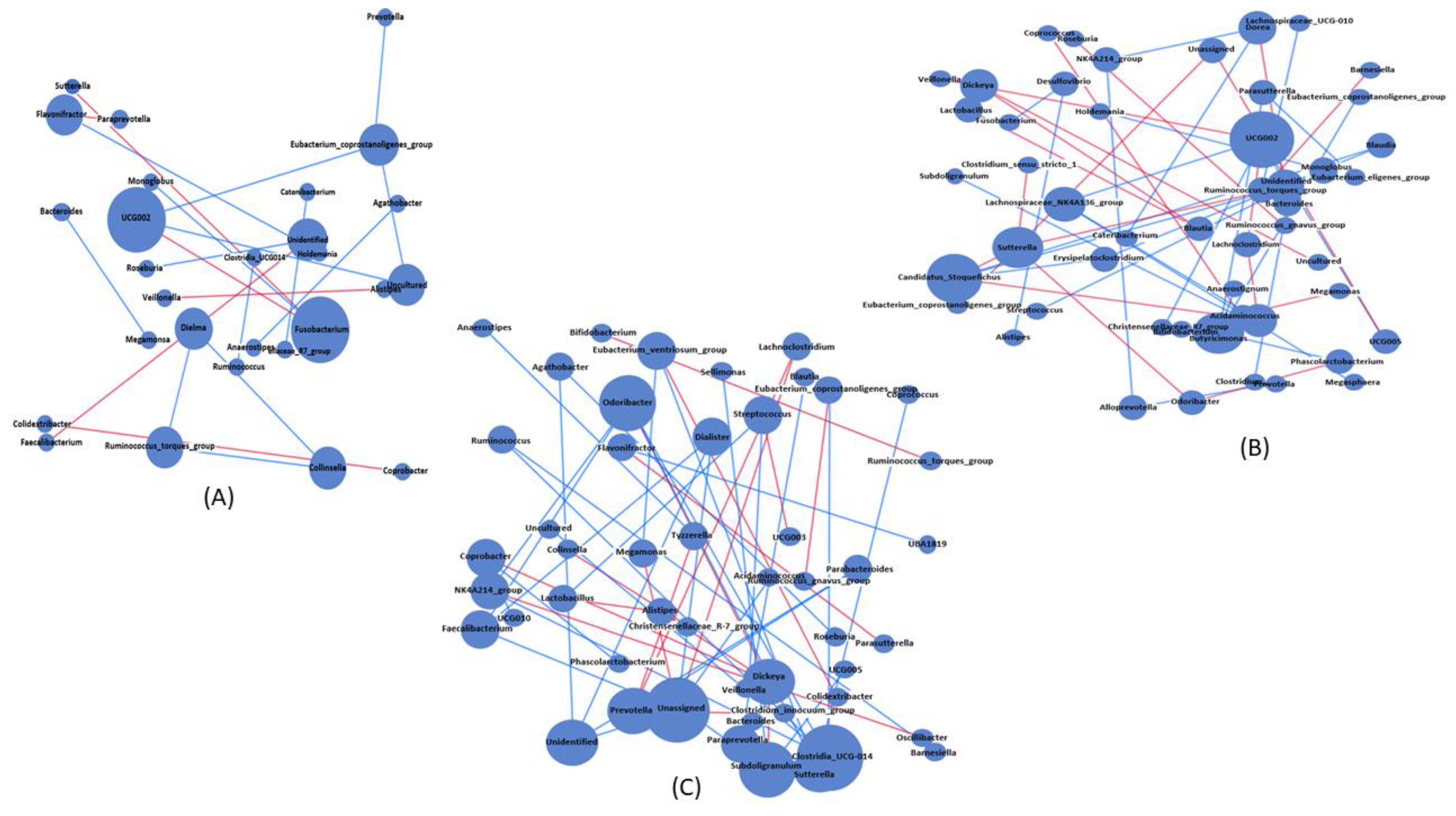
3.5. Bacterial Association and Interaction Analysis among the Experimental Groups
3.6. Predicted Functional Shift among the Experimental Groups
3.7. Correlation and Association Analyses among the Significant Taxa, Predicted Pathways, and Physiological Parameters
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rettedal, E.A.; Ilesanmi-Oyelere, B.L.; Roy, N.C.; Coad, J.; Kruger, M.C. The Gut Microbiome Is Altered in Postmenopausal Women with Osteoporosis and Osteopenia. JBMR Plus 2021, 5, e10452. [Google Scholar] [CrossRef] [PubMed]
- Raftar, S.K.A.; Tavassol, Z.H.; Amiri, M.; Ejtahed, H.-S.; Zangeneh, M.; Sadeghi, S.; Ashrafian, F.; Kariman, A.; Khatami, S.; Siadat, S.D. Assessment of fecal Akkermansia muciniphila in patients with osteoporosis and osteopenia: A pilot study. J. Diabetes Metab. Disord. 2021, 20, 279–284. [Google Scholar] [CrossRef] [PubMed]
- Siris, E.S.; Simon, J.A.; Barton, I.P.; McClung, M.R.; Grauer, A. Effects of risedronate on fracture risk in postmenopausal women with osteopenia. Osteoporos. Int. 2008, 19, 681–686. [Google Scholar] [CrossRef] [Green Version]
- Ma, S.; Qin, J.; Hao, Y.; Fu, L. Association of gut microbiota composition and function with an aged rat model of senile osteoporosis using 16S rRNA and metagenomic sequencing analysis. Aging 2020, 12, 10795–10808. [Google Scholar] [CrossRef] [PubMed]
- Aziziyeh, R.; Amin, M.; Habib, M.; Perlaza, J.G.; Szafranski, K.; McTavish, R.K.; Disher, T.; Lüdke, A.; Cameron, C. The burden of osteoporosis in four Latin American countries: Brazil, Mexico, Colombia, and Argentina. J. Med. Econ. 2019, 22, 638–644. [Google Scholar] [CrossRef] [PubMed]
- Fischer, S.; Kapinos, K.; Mulcahy, A.; Pinto, L.; Hayden, O.; Barron, R. Estimating the long-term functional burden of osteoporosis-related fractures. Osteoporos. Int. 2017, 28, 2843–2851. [Google Scholar] [CrossRef]
- Chen, X.; Wang, Z.; Duan, N.; Zhu, G.; Schwarz, E.M.; Xie, C. Osteoblast–osteoclast interactions. Connect. Tissue Res. 2018, 59, 99–107. [Google Scholar] [CrossRef]
- Seely, K.D.; Kotelko, C.A.; Douglas, H.; Bealer, B.; Brooks, A.E. The Human Gut Microbiota: A Key Mediator of Osteoporosis and Osteogenesis. Int. J. Mol. Sci. 2021, 22, 9452. [Google Scholar] [CrossRef]
- He, J.; Xu, S.; Zhang, B.; Xiao, C.; Chen, Z.; Si, F.; Fu, J.; Lin, X.; Zheng, G.; Yu, G.; et al. Gut microbiota and metabolite alterations associated with reduced bone mineral density or bone metabolic indexes in postmenopausal osteoporosis. Aging 2020, 12, 8583–8604. [Google Scholar] [CrossRef]
- Bijelic, R.; Milicevic, S.; Balaban, J. Risk Factors for Osteoporosis in Postmenopausal Women. Med. Arch. 2017, 71, 25–28. [Google Scholar] [CrossRef]
- Wang, J.; Wang, Y.; Gao, W.; Wang, B.; Zhao, H.; Zeng, Y.; Ji, Y.; Hao, D. Diversity analysis of gut microbiota in osteoporosis and osteopenia patients. PeerJ 2017, 5, e3450. [Google Scholar] [CrossRef] [Green Version]
- Sjögren, K.; Engdahl, C.; Henning, P.; Lerner, U.H.; Tremaroli, V.; Lagerquist, M.K.; Bäckhed, F.; Ohlsson, C. The gut microbiota regulates bone mass in mice. J. Bone Miner. Res. 2012, 27, 1357–1367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scholz-Ahrens, K.E.; Ade, P.; Marten, B.; Weber, P.; Timm, W.; Aςil, Y.; Glüer, C.-C.; Schrezenmeir, J. Prebiotics, Probiotics, and Synbiotics Affect Mineral Absorption, Bone Mineral Content, and Bone Structure. J. Nutr. 2007, 137, 838S–846S. [Google Scholar] [CrossRef] [PubMed]
- Clarke, G.; Stilling, R.M.; Kennedy, P.J.; Stanton, C.; Cryan, J.F.; Dinan, T.G. Minireview: Gut microbiota: The neglected endocrine organ. Mol. Endocrinol. 2014, 28, 1221–1238. [Google Scholar] [CrossRef] [Green Version]
- Daniel, N.; Lécuyer, E.; Chassaing, B. Host/microbiota interactions in health and diseases—Time for mucosal microbiology! Mucosal Immunol. 2021, 14, 1006–1016. [Google Scholar] [CrossRef]
- Fang, C.-Y.; Chen, J.-S.; Hsu, B.-M.; Hussain, B.; Rathod, J.; Lee, K.-H. Colorectal Cancer Stage-Specific Fecal Bacterial Community Fingerprinting of the Taiwanese Population and Underpinning of Potential Taxonomic Biomarkers. Microorganisms 2021, 9, 1548. [Google Scholar] [CrossRef] [PubMed]
- Loftus, M.; Hassouneh, S.A.-D.; Yooseph, S. Bacterial associations in the healthy human gut microbiome across populations. Sci. Rep. 2021, 11, 2828. [Google Scholar] [CrossRef] [PubMed]
- Kuntal, B.K.; Chandrakar, P.; Sadhu, S.; Mande, S.S. ‘NetShift’: A methodology for understanding ‘driver microbes’ from healthy and disease microbiome datasets. ISME J. 2019, 13, 442–454. [Google Scholar] [CrossRef] [Green Version]
- De Matos, O.; Da Silva, D.J.L.; de Oliveira, J.M.; Castelo-Branco, C. Effect of specific exercise training on bone mineral density in women with postmenopausal osteopenia or osteoporosis. Gynecol. Endocrinol. 2009, 25, 616–620. [Google Scholar] [CrossRef]
- Xu, Z.; Xie, Z.; Sun, J.; Huang, S.; Chen, Y.; Li, C.; Sun, X.; Xia, B.; Tian, L.; Guo, C.; et al. Gut Microbiome Reveals Specific Dysbiosis in Primary Osteoporosis. Front. Cell. Infect. Microbiol. 2020, 10, 160. [Google Scholar] [CrossRef]
- Yan, J.; Herzog, J.W.; Tsang, K.; Brennan, C.A.; Bower, M.A.; Garrett, W.S.; Sartor, B.R.; Aliprantis, A.O.; Charles, J.F. Gut microbiota induce IGF-1 and promote bone formation and growth. Proc. Natl. Acad. Sci. USA 2016, 113, E7554–E7563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ling, C.-W.; Miao, Z.; Xiao, M.-L.; Zhou, H.; Jiang, Z.; Fu, Y.; Xiong, F.; Zuo, L.; Liu, Y.-P.; Wu, Y.-Y.; et al. The Association between Gut Microbiota and Osteoporosis was Mediated by Amino Acid Metabolism: Multi-omics Integration in a Large Adult Cohort. medRxiv 2020, medRxiv:2020.08.28.20183764. [Google Scholar] [CrossRef]
- Li, C.; Huang, Q.; Yang, R.; Dai, Y.; Zeng, Y.; Tao, L.; Li, X.; Zeng, J.; Wang, Q. Gut microbiota composition and bone mineral loss—Epidemiologic evidence from individuals in Wuhan, China. Osteoporos. Int. 2019, 30, 1003–1013. [Google Scholar] [CrossRef] [PubMed]
- The Writing Group for the ISCD Position Development Conference. Indications and Reporting for Dual-Energy X-ray Absorptiometry. J. Clin. Densitom. 2004, 7, 37–44. [Google Scholar] [CrossRef]
- Lim, M.Y.; Song, E.-J.; Kim, S.H.; Lee, J.; Nam, Y.-D. Comparison of DNA extraction methods for human gut microbial community profiling. Syst. Appl. Microbiol. 2018, 41, 151–157. [Google Scholar] [CrossRef]
- Huang, T.-Y.; Hsu, B.-M.; Chao, W.-C.; Fan, C.-W. Plant n-alkane production from litterfall altered the diversity and community structure of alkane degrading bacteria in litter layer in lowland subtropical rainforest in Taiwan. Biogeosciences 2018, 15, 1815–1826. [Google Scholar] [CrossRef] [Green Version]
- Nagpal, S.; Singh, R.; Yadav, D.; Mande, S.S. MetagenoNets: Comprehensive inference and meta-insights for microbial correlation networks. Nucleic Acids Res. 2020, 48, W572–W579. [Google Scholar] [CrossRef]
- Douglas, G.M.; Maffei, V.J.; Zaneveld, J.; Yurgel, S.N.; Brown, J.R.; Taylor, C.M.; Huttenhower, C.; Langille, M.G.I. PIC-RUSt2: An improved and customizable approach for metagenome inference. bioRxiv 2020, bioRxiv:672295. [Google Scholar] [CrossRef] [Green Version]
- Das, M.; Cronin, O.; Keohane, D.M.; Cormac, E.M.; Nugent, H.; Nugent, M.; Molloy, C.; O’Toole, P.W.; Shanahan, F.; Molloy, M.G.; et al. Gut microbiota alterations associated with reduced bone mineral density in older adults. Rheumatology 2019, 58, 2295–2304. [Google Scholar] [CrossRef] [Green Version]
- Ibáñez, L.; Rouleau, M.; Wakkach, A.; Blin-Wakkach, C. Gut microbiome and bone. Jt. Bone Spine 2019, 86, 43–47. [Google Scholar] [CrossRef]
- Li, J.; Ho, W.T.P.; Liu, C.; Chow, S.K.-H.; Ip, M.; Yu, J.; Wong, H.S.; Cheung, W.-H.; Sung, J.J.Y.; Wong, R.M.Y. The role of gut microbiota in bone homeostasis. Bone Jt. Res. 2021, 10, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Wei, Q.; Xu, D.; Wei, Y.; Wang, J.; Amakye, W.K.; Pan, J.; Cui, Z.; Zhang, Z. The associations of gut microbiota and fecal short-chain fatty acids with bone mass were largely mediated by weight status: A cross-sectional study. Eur. J. Nutr. 2021, 60, 4505–4517. [Google Scholar] [CrossRef] [PubMed]
- Di Stefano, M.; Veneto, G.; Malservisi, S.; Corazza, G.R. Small Intestine Bacterial Overgrowth and Metabolic Bone Disease. Dig. Dis. Sci. 2001, 46, 1077–1082. [Google Scholar] [CrossRef] [PubMed]
- Stotzer, P.-O.; Johansson, C.; Mellström, D.; Lindstedt, G.; Kilander, A.F. Bone mineral density in patients with small intestinal bacterial overgrowth. Hepato-Gastroenterology 2003, 50, 1415–1418. [Google Scholar]
- Kelder, T.; Stroeve, J.H.M.; Bijlsma, S.; Radonjić, M.; Roeselers, G. Correlation network analysis reveals relationships between diet-induced changes in human gut microbiota and metabolic health. Nutr. Diabetes 2014, 4, e122. [Google Scholar] [CrossRef] [Green Version]
- Gaike, A.H.; Paul, D.; Bhute, S.; Dhotre, D.P.; Pande, P.; Upadhyaya, S.; Reddy, Y.; Sampath, R.; Ghosh, D.; Chandraprabha, D.; et al. The Gut Microbial Diversity of Newly Diagnosed Diabetics but not of Prediabetics Is Significantly Different from That of Healthy Nondiabetics. Msystems 2020, 5, e00578-19. [Google Scholar] [CrossRef] [Green Version]
- Kang, Y.; Feng, D.; Law, H.K.-W.; Qu, W.; Wu, Y.; Zhu, G.-H.; Huang, W.-Y. Compositional alterations of gut microbiota in children with primary nephrotic syndrome after initial therapy. BMC Nephrol. 2019, 20, 434. [Google Scholar] [CrossRef]
- Aslam, M.N.; Bassis, C.M.; Zhang, L.; Zaidi, S.; Varani, J.; Bergin, I.L. Calcium Reduces Liver Injury in Mice on a High-Fat Diet: Alterations in Microbial and Bile Acid Profiles. PLoS ONE 2016, 11, e0166178. [Google Scholar] [CrossRef]
- Rosero, J.A.; Killer, J.; Sechovcová, H.; Mrázek, J.; Benada, O.; Fliegerová, K.; Havlík, J.; Kopečný, J. Reclassification of Eubacterium rectale (Hauduroy et al. 1937) Prévot 1938 in a new genus Agathobacter gen. nov. as Agathobacter rectalis comb. nov., and description of Agathobacter ruminis sp. nov., isolated from the rumen contents of sheep and cows. Int. J. Syst. Evol. Microbiol. 2016, 66, 768–773. [Google Scholar] [CrossRef]
- Hua, X.; Zhu, J.; Yang, T.; Guo, M.; Li, Q.; Chen, J.; Li, T. The Gut Microbiota and Associated Metabolites Are Altered in Sleep Disorder of Children with Autism Spectrum Disorders. Front. Psychiatry 2020, 11, 855. [Google Scholar] [CrossRef]
- Tyagi, A.M.; Yu, M.; Darby, T.M.; Vaccaro, C.; Li, J.-Y.; Owens, J.A.; Hsu, E.; Adams, J.; Weitzmann, M.N.; Jones, R.M.; et al. The Microbial Metabolite Butyrate Stimulates Bone Formation via T Regulatory Cell-Mediated Regulation of WNT10B Expression. Immunity 2018, 49, 1116–1131.e7. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Boer, C.G.; Oei, L.; Medina-Gomez, C. The Gut Microbiome: A New Frontier in Musculoskeletal Research. Curr. Osteoporos. Rep. 2021, 19, 347–357. [Google Scholar] [CrossRef] [PubMed]
- Atarashi, K.; Tanoue, T.; Shima, T.; Imaoka, A.; Kuwahara, T.; Momose, Y.; Cheng, G.; Yamasaki, S.; Saito, T.; Ohba, Y.; et al. Induction of colonic regulatory T cells by indigenous clostridium species. Science 2011, 331, 337–341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Martinis, M.; Ginaldi, L.; Allegra, A.; Sirufo, M.M.; Pioggia, G.; Tonacci, A.; Gangemi, S. The Osteoporosis/Microbiota Linkage: The Role of miRNA. Int. J. Mol. Sci. 2020, 21, 8887. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Charles, J.F. Gut Microbiome and Bone: To Build, Destroy, or Both? Curr. Osteoporos. Rep. 2017, 15, 376–384. [Google Scholar] [CrossRef]
- Caspi, R.; Billington, R.; Fulcher, C.A.; Keseler, I.M.; Kothari, A.; Krummenacker, M.; Latendresse, M.; Midford, P.E.; Ong, Q.; Ong, W.K.; et al. The MetaCyc database of metabolic pathways and enzymes. Nucleic Acids Res. 2017, 46, D633–D639. [Google Scholar] [CrossRef] [Green Version]
- Dröge, W. Free Radicals in the Physiological Control of Cell Function. Physiol. Rev. 2002, 82, 47–95. [Google Scholar] [CrossRef] [Green Version]
- Callaway, D.A.; Jiang, J.X. Reactive oxygen species and oxidative stress in osteoclastogenesis, skeletal aging and bone diseases. J. Bone Miner. Metab. 2015, 33, 359–370. [Google Scholar] [CrossRef]
- Garrett, I.; Gutierrez, G.; Mundy, G. Statins and bone formation. Curr. Pharm. Des. 2001, 7, 715–736. [Google Scholar] [CrossRef]
- Hasan, W.N.; Chin, K.Y.; Jolly, J.J.; Ghafar, N.A.; Soelaiman, I.N. Identifying potential therapeutics for osteoporosis by exploiting the relationship between mevalonate pathway and bone metabolism. Endocr. Metab. Immune Disord. Drug Targets (Former. Curr. Drug Targets-Immune Endocr. Metab. Disord.) 2018, 18, 450–457. [Google Scholar] [CrossRef]
- Kim, B.; Cho, Y.J.; Lim, W. Osteoporosis therapies and their mechanisms of action (Review). Exp. Ther. Med. 2021, 22, 1379. [Google Scholar] [CrossRef] [PubMed]
- Øyen, J.; Svingen, G.F.T.; Gjesdal, C.G.; Tell, G.S.; Ueland, P.M.; Lysne, V.; Apalset, E.M.; Meyer, K.; Vollset, S.E.; Nygård, O.K. Plasma dimethylglycine, nicotine exposure and risk of low bone mineral density and hip fracture: The Hordaland Health Study. Osteoporos. Int. 2015, 26, 1573–1583. [Google Scholar] [CrossRef] [PubMed]
- Farooqi, I.N.; Sharma, A. Unusual Case of Pelvic Osteomyelitis Revealing Osteoporosis. J. Endocr. Soc. 2021, 5, A230–A231. [Google Scholar] [CrossRef]
- Zhu, C.; Liu, J.; Pan, Y.; Fan, Y.; Jin, L.; Liu, Y.; Zhang, Z.; Gan, Y.; Tang, W.; Li, J.; et al. Alteration of fecal microbial compositions and bacterial taxa in female osteoporotic patients. bioRxiv 2020, bioRxiv:2020.2001.2021.914903. [Google Scholar] [CrossRef]
- Cheng, S.; Qi, X.; Ma, M.; Zhang, L.; Cheng, B.; Liang, C.; Liu, L.; Li, P.; Kafle, O.P.; Wen, Y.; et al. Assessing the Relationship Between Gut Microbiota and Bone Mineral Density. Front. Genet. 2020, 11, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, M.; Li, C.; Dai, Y.; Zhou, H.; Cui, Y.; Zeng, Y.; Huang, Q.; Wang, Q. High-Throughput Absolute Quantification Sequencing Revealed Osteoporosis-Related Gut Microbiota Alterations in Han Chinese Elderly. Front. Cell. Infect. Microbiol. 2021, 11, 630372. [Google Scholar] [CrossRef]
- Xiao, S.; Fei, N.; Pang, X.; Shen, J.; Wang, L.; Zhang, B.; Zhang, M.; Zhang, X.; Zhang, C.; Li, M.; et al. A gut microbiota-targeted dietary intervention for amelioration of chronic inflammation underlying metabolic syndrome. FEMS Microbiol. Ecol. 2014, 87, 357–367. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kuo, Y.-J.; Chen, C.-J.; Hussain, B.; Tsai, H.-C.; Hsu, G.-J.; Chen, J.-S.; Asif, A.; Fan, C.-W.; Hsu, B.-M. Inferring Bacterial Community Interactions and Functionalities Associated with Osteopenia and Osteoporosis in Taiwanese Postmenopausal Women. Microorganisms 2023, 11, 234. https://doi.org/10.3390/microorganisms11020234
Kuo Y-J, Chen C-J, Hussain B, Tsai H-C, Hsu G-J, Chen J-S, Asif A, Fan C-W, Hsu B-M. Inferring Bacterial Community Interactions and Functionalities Associated with Osteopenia and Osteoporosis in Taiwanese Postmenopausal Women. Microorganisms. 2023; 11(2):234. https://doi.org/10.3390/microorganisms11020234
Chicago/Turabian StyleKuo, Yi-Jie, Chia-Jung Chen, Bashir Hussain, Hsin-Chi Tsai, Gwo-Jong Hsu, Jung-Sheng Chen, Aslia Asif, Cheng-Wei Fan, and Bing-Mu Hsu. 2023. "Inferring Bacterial Community Interactions and Functionalities Associated with Osteopenia and Osteoporosis in Taiwanese Postmenopausal Women" Microorganisms 11, no. 2: 234. https://doi.org/10.3390/microorganisms11020234