
The Role of Gut Microbiota-Derived Lithocholic Acid, Deoxycholic Acid and Their Derivatives on the Function and Differentiation of Immune Cells

Abstract
:1. Introduction
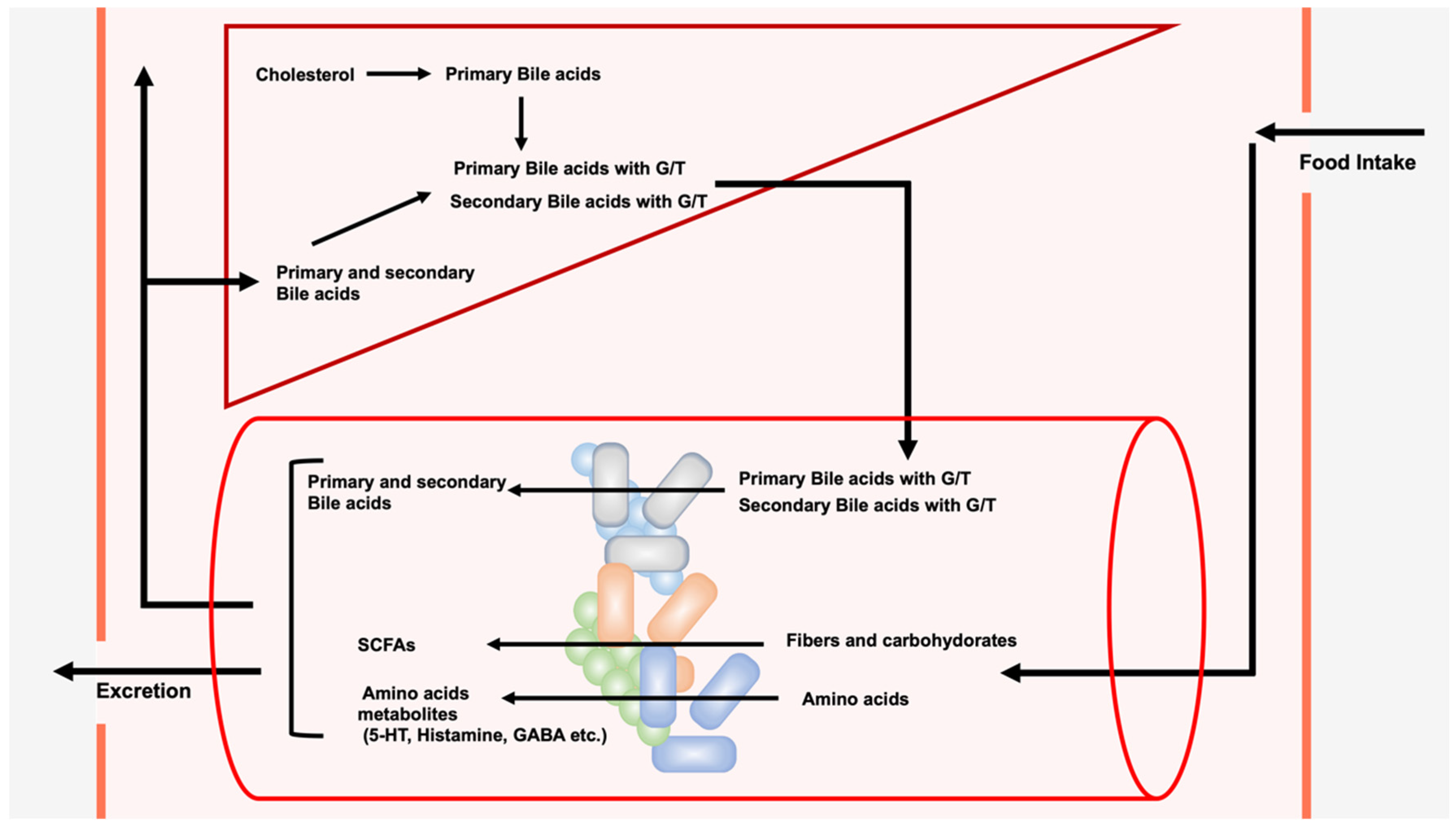
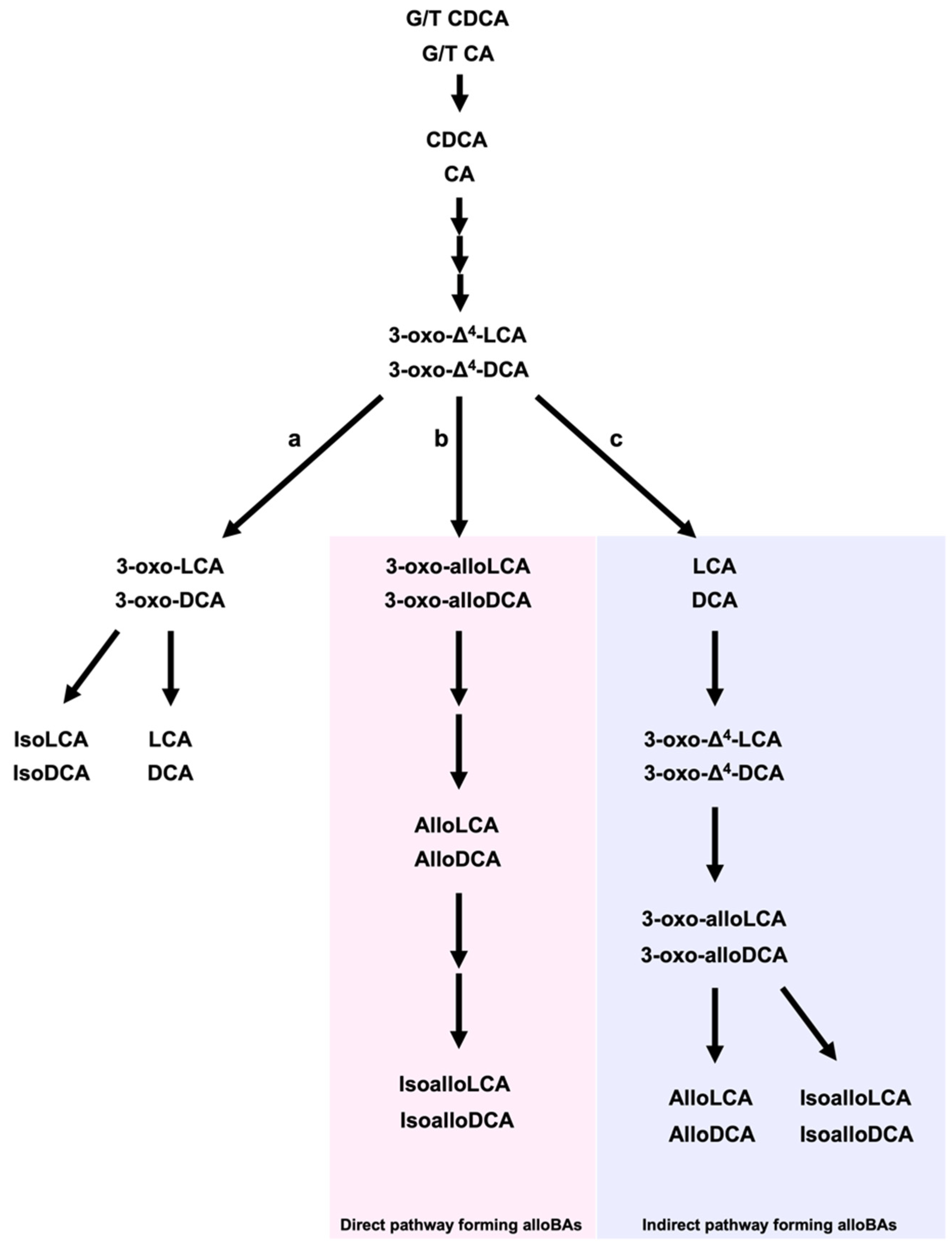
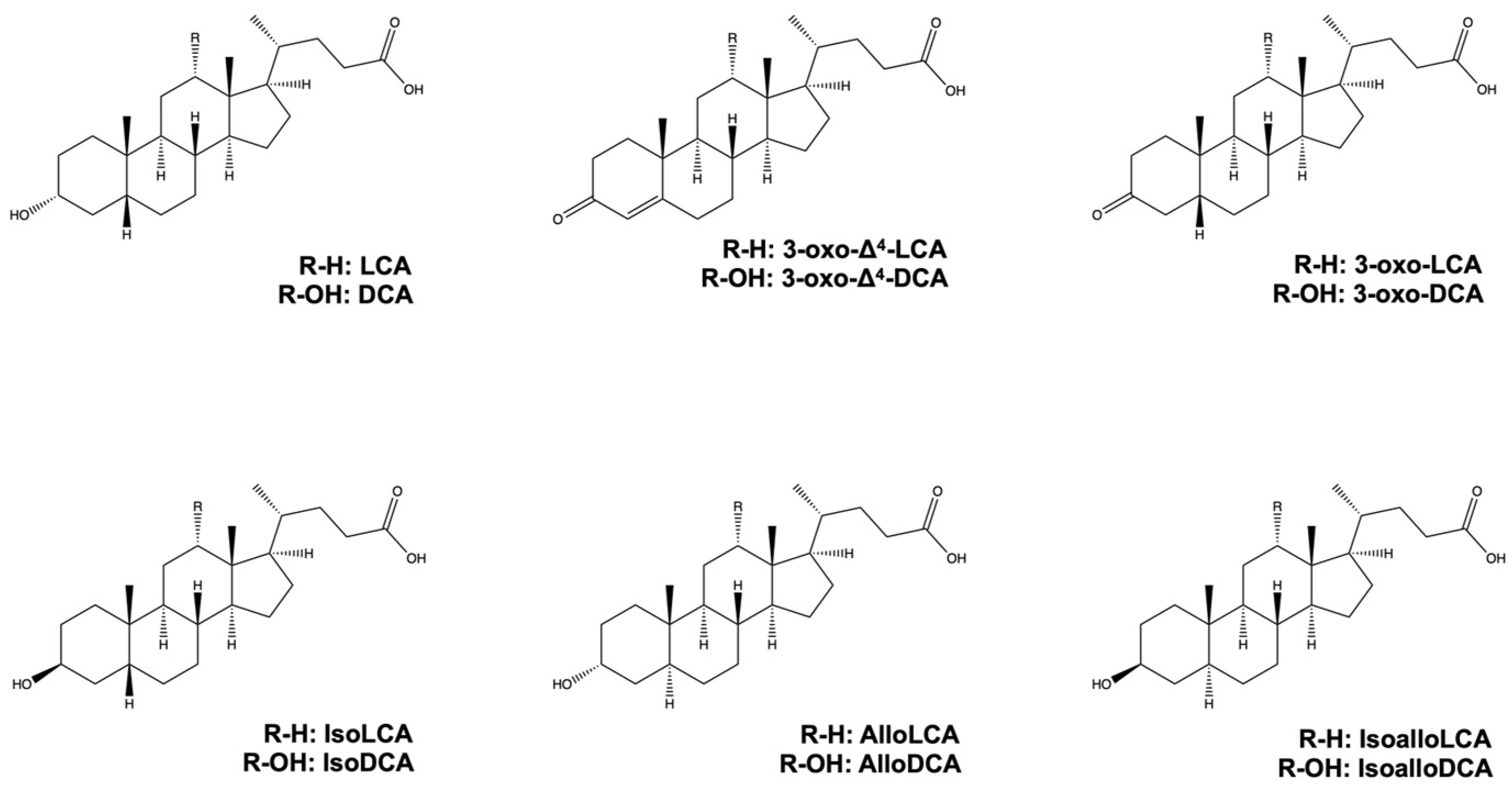
2. LCA, DCA, and Their Derivatives from Gut Microbiota
3. Role of LCA, DCA, and Their Derivatives on Immune cells
3.1. Antigen-Presenting Cells and Helper T Cell Subset
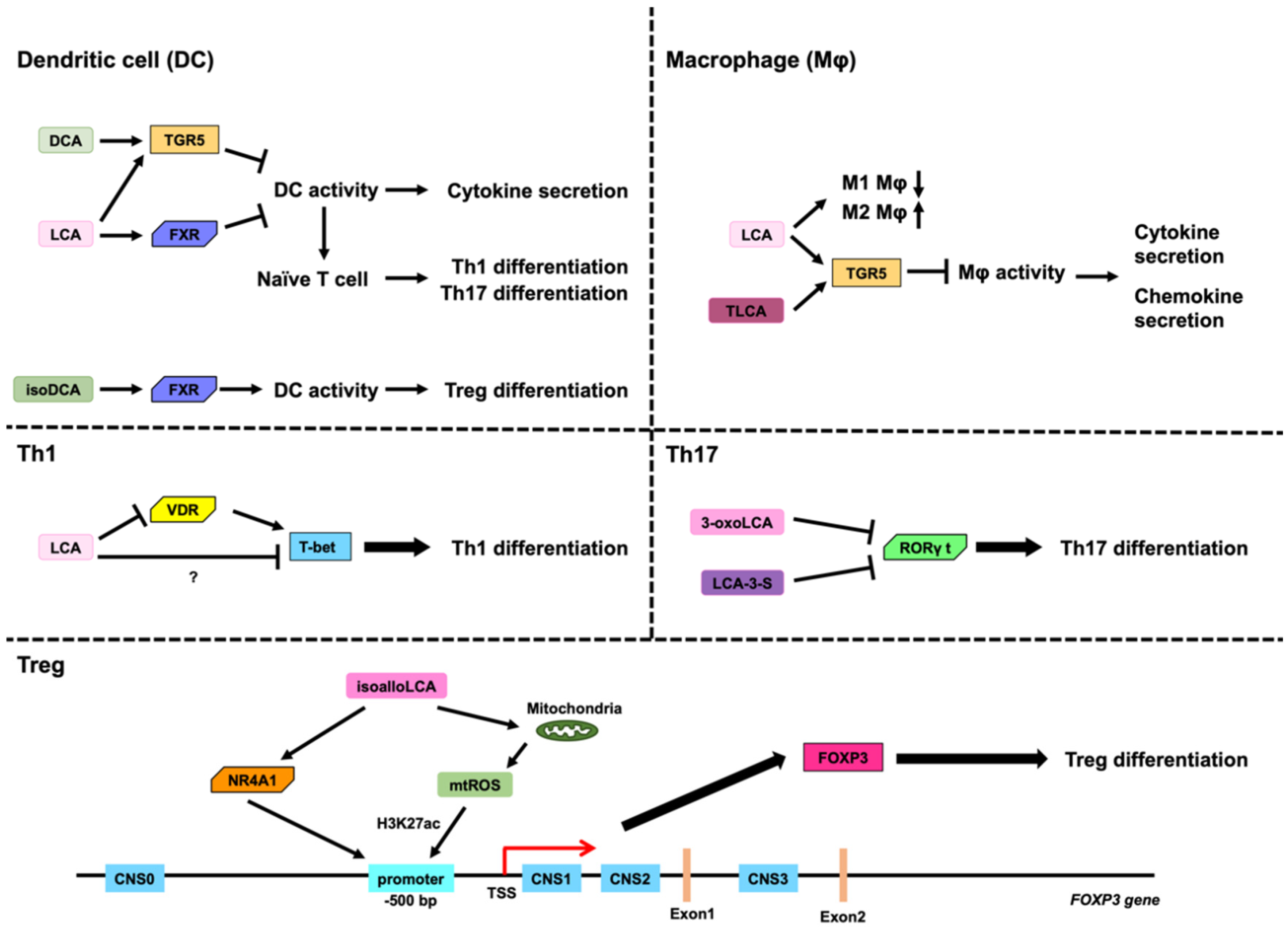
3.2. Role of LCA, DCA, and Their Derivatives on DCs
3.3. Role of LCA, DCA, and Their Derivatives on Macropahges
3.4. Role of LCA and DCA on Th1 Cells
3.5. Role of LCA, DCA, and Their Derivatives on Th17 Cells
3.6. Role of LCA, DCA, and Their Derivatives on Treg Cells
4. Concluding Remarks
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Sender, R.; Fuchs, S.; Milo, R. Revised Estimates for the Number of Human and Bacteria Cells in the Body. PLoS Biol. 2016, 14, e1002533. [Google Scholar] [CrossRef]
- Dekaboruah, E.; Suryavanshi, M.V.; Chettri, D.; Verma, A.K. Human microbiome: An academic update on human body site specific surveillance and its possible role. Arch. Microbiol. 2020, 202, 2147–2167. [Google Scholar] [CrossRef]
- Donaldson, G.P.; Lee, S.M.; Mazmanian, S.K. Gut biogeography of the bacterial microbiota. Nat. Rev. Microbiol. 2016, 14, 20–32. [Google Scholar] [CrossRef]
- Mishra, K.; Bukavina, L.; Ghannoum, M. Symbiosis and Dysbiosis of the Human Mycobiome. Front. Microbiol. 2021, 12, 636131. [Google Scholar] [CrossRef]
- Hou, K.; Wu, Z.X.; Chen, X.Y.; Wang, J.Q.; Zhang, D.; Xiao, C.; Zhu, D.; Koya, J.B.; Wei, L.; Li, J.; et al. Microbiota in health and diseases. Signal Transduct. Target Ther. 2022, 7, 135. [Google Scholar] [CrossRef]
- Zhao, M.; Chu, J.; Feng, S.; Guo, C.; Xue, B.; He, K.; Li, L. Immunological mechanisms of inflammatory diseases caused by gut microbiota dysbiosis: A review. Biomed. Pharmacother. 2023, 164, 114985. [Google Scholar] [CrossRef]
- Liu, Q.; Su, Q.; Zhang, F.; Tun, H.M.; Mak, J.W.Y.; Lui, G.C.; Ng, S.S.S.; Ching, J.Y.L.; Li, A.; Lu, W.; et al. Multi-kingdom gut microbiota analyses define COVID-19 severity and post-acute COVID-19 syndrome. Nat. Commun. 2022, 13, 6806. [Google Scholar] [CrossRef]
- Zhang, F.; Lau, R.I.; Liu, Q.; Su, Q.; Chan, F.K.L.; Ng, S.C. Gut microbiota in COVID-19: Key microbial changes, potential mechanisms and clinical applications. Nat. Rev. Gastroenterol. Hepatol. 2023, 20, 323–337. [Google Scholar] [CrossRef]
- Wang, B.; Zhang, L.; Wang, Y.; Dai, T.; Qin, Z.; Zhou, F.; Zhang, L. Alterations in microbiota of patients with COVID-19: Potential mechanisms and therapeutic interventions. Signal Transduct. Target Ther. 2022, 7, 143. [Google Scholar] [CrossRef]
- Van der Hee, B.; Wells, J.M. Microbial Regulation of Host Physiology by Short-chain Fatty Acids. Trends Microbiol. 2021, 29, 700–712. [Google Scholar] [CrossRef]
- Portincasa, P.; Bonfrate, L.; Vacca, M.; De Angelis, M.; Farella, I.; Lanza, E.; Khalil, M.; Wang, D.Q.; Sperandio, M.; Di Ciaula, A. Gut Microbiota and Short Chain Fatty Acids: Implications in Glucose Homeostasis. Int. J. Mol. Sci. 2022, 23, 1105. [Google Scholar] [CrossRef] [PubMed]
- Jameson, K.G.; Olson, C.A.; Kazmi, S.A.; Hsiao, E.Y. Toward Understanding Microbiome-Neuronal Signaling. Mol. Cell 2020, 78, 577–583. [Google Scholar] [CrossRef]
- Sun, P.; Su, L.; Zhu, H.; Li, X.; Guo, Y.; Du, X.; Zhang, L.; Qin, C. Gut Microbiota Regulation and Their Implication in the Development of Neurodegenerative Disease. Microorganisms 2021, 9, 2281. [Google Scholar] [CrossRef] [PubMed]
- Kiriyama, Y.; Nochi, H. The Biosynthesis, Signaling, and Neurological Functions of Bile Acids. Biomolecules 2019, 9, 232. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Dawson, P.A. Animal models to study bile acid metabolism. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 895–911. [Google Scholar] [CrossRef] [PubMed]
- Straniero, S.; Laskar, A.; Savva, C.; Hardfeldt, J.; Angelin, B.; Rudling, M. Of mice and men: Murine bile acids explain species differences in the regulation of bile acid and cholesterol metabolism. J. Lipid Res. 2020, 61, 480–491. [Google Scholar] [CrossRef]
- Sun, J.; Li, M.; Zhou, H.; Chong, J.; Zhang, J.; Yu, B.; Chen, D.; Ge, L. Importance of gut microbiota for bile acid composition and concentration in pigs. Front. Anim. Sci. 2022, 3, 951840. [Google Scholar] [CrossRef]
- Kiriyama, Y.; Nochi, H. Physiological Role of Bile Acids Modified by the Gut Microbiome. Microorganisms 2021, 10, 68. [Google Scholar] [CrossRef]
- Lee, J.W.; Cowley, E.S.; Wolf, P.G.; Doden, H.L.; Murai, T.; Caicedo, K.Y.O.; Ly, L.K.; Sun, F.; Takei, H.; Nittono, H.; et al. Formation of secondary allo-bile acids by novel enzymes from gut Firmicutes. Gut. Microbes 2022, 14, 2132903. [Google Scholar] [CrossRef]
- Doden, H.L.; Ridlon, J.M. Microbial Hydroxysteroid Dehydrogenases: From Alpha to Omega. Microorganisms 2021, 9, 469. [Google Scholar] [CrossRef]
- Kiriyama, Y.; Nochi, H. Role of Microbiota-Modified Bile Acids in the Regulation of Intracellular Organelles and Neurodegenerative Diseases. Genes 2023, 14, 825. [Google Scholar] [CrossRef]
- Che, Y.; Xu, W.; Ding, C.; He, T.; Xu, X.; Shuai, Y.; Huang, H.; Wu, J.; Wang, Y.; Wang, C.; et al. Bile acids target mitofusin 2 to differentially regulate innate immunity in physiological versus cholestatic conditions. Cell Rep. 2023, 42, 112011. [Google Scholar] [CrossRef]
- Jia, W.; Xie, G.; Jia, W. Bile acid-microbiota crosstalk in gastrointestinal inflammation and carcinogenesis. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 111–128. [Google Scholar] [CrossRef] [PubMed]
- Campbell, C.; McKenney, P.T.; Konstantinovsky, D.; Isaeva, O.I.; Schizas, M.; Verter, J.; Mai, C.; Jin, W.B.; Guo, C.J.; Violante, S.; et al. Bacterial metabolism of bile acids promotes generation of peripheral regulatory T cells. Nature 2020, 581, 475–479. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Xie, S.; Chi, Z.; Zhang, J.; Liu, Y.; Zhang, L.; Zheng, M.; Zhang, X.; Xia, D.; Ke, Y.; et al. Bile Acids Control Inflammation and Metabolic Disorder through Inhibition of NLRP3 Inflammasome. Immunity 2016, 45, 802–816. [Google Scholar] [CrossRef] [PubMed]
- Hang, S.; Paik, D.; Yao, L.; Kim, E.; Trinath, J.; Lu, J.; Ha, S.; Nelson, B.N.; Kelly, S.P.; Wu, L.; et al. Bile acid metabolites control T(H)17 and T(reg) cell differentiation. Nature 2019, 576, 143–148. [Google Scholar] [CrossRef]
- Hu, J.; Wang, C.; Huang, X.; Yi, S.; Pan, S.; Zhang, Y.; Yuan, G.; Cao, Q.; Ye, X.; Li, H. Gut microbiota-mediated secondary bile acids regulate dendritic cells to attenuate autoimmune uveitis through TGR5 signaling. Cell Rep. 2021, 36, 109726. [Google Scholar] [CrossRef]
- Hu, J.; Zhang, Y.; Yi, S.; Wang, C.; Huang, X.; Pan, S.; Yang, J.; Yuan, G.; Tan, S.; Li, H. Lithocholic acid inhibits dendritic cell activation by reducing intracellular glutathione via TGR5 signaling. Int. J. Biol. Sci. 2022, 18, 4545–4559. [Google Scholar] [CrossRef]
- Li, W.; Hang, S.; Fang, Y.; Bae, S.; Zhang, Y.; Zhang, M.; Wang, G.; McCurry, M.D.; Bae, M.; Paik, D.; et al. A bacterial bile acid metabolite modulates T(reg) activity through the nuclear hormone receptor NR4A1. Cell Host Microbe 2021, 29, 1366–1377.e9. [Google Scholar] [CrossRef]
- Roggeri, A.; Schepers, M.; Tiane, A.; Rombaut, B.; van Veggel, L.; Hellings, N.; Prickaerts, J.; Pittaluga, A.; Vanmierlo, T. Sphingosine-1-Phosphate Receptor Modulators and Oligodendroglial Cells: Beyond Immunomodulation. Int. J. Mol. Sci. 2020, 21, 7537. [Google Scholar] [CrossRef]
- Pandak, W.M.; Kakiyama, G. The acidic pathway of bile acid synthesis: Not just an alternative pathway. Liver Res. 2019, 3, 88–98. [Google Scholar] [CrossRef] [PubMed]
- Russell, D.W. Fifty years of advances in bile acid synthesis and metabolism. J. Lipid Res. 2009, 50, S120–S125. [Google Scholar] [CrossRef] [PubMed]
- Chiang, J.Y.L.; Ferrell, J.M. Up to date on cholesterol 7 alpha-hydroxylase (CYP7A1) in bile acid synthesis. Liver Res. 2020, 4, 47–63. [Google Scholar] [CrossRef] [PubMed]
- Chiang, J.Y.L.; Ferrell, J.M. Bile Acids as Metabolic Regulators and Nutrient Sensors. Annu. Rev. Nutr. 2019, 39, 175–200. [Google Scholar] [CrossRef]
- Xue, R.; Su, L.; Lai, S.; Wang, Y.; Zhao, D.; Fan, J.; Chen, W.; Hylemon, P.B.; Zhou, H. Bile Acid Receptors and the Gut-Liver Axis in Nonalcoholic Fatty Liver Disease. Cells 2021, 10, 2806. [Google Scholar] [CrossRef]
- Kenna, J.G.; Taskar, K.S.; Battista, C.; Bourdet, D.L.; Brouwer, K.L.R.; Brouwer, K.R.; Dai, D.; Funk, C.; Hafey, M.J.; Lai, Y.; et al. Can Bile Salt Export Pump Inhibition Testing in Drug Discovery and Development Reduce Liver Injury Risk? An International Transporter Consortium Perspective. Clin. Pharmacol. Ther. 2018, 104, 916–932. [Google Scholar] [CrossRef]
- Daly, J.W.; Keely, S.J.; Gahan, C.G.M. Functional and Phylogenetic Diversity of BSH and PVA Enzymes. Microorganisms 2021, 9, 732. [Google Scholar] [CrossRef]
- Jones, B.V.; Begley, M.; Hill, C.; Gahan, C.G.; Marchesi, J.R. Functional and comparative metagenomic analysis of bile salt hydrolase activity in the human gut microbiome. Proc. Natl. Acad. Sci. USA 2008, 105, 13580–13585. [Google Scholar] [CrossRef]
- O’Flaherty, S.; Briner Crawley, A.; Theriot, C.M.; Barrangou, R. The Lactobacillus Bile Salt Hydrolase Repertoire Reveals Niche-Specific Adaptation. mSphere 2018, 3, 10–1128. [Google Scholar] [CrossRef]
- Clarke, G.; Sandhu, K.V.; Griffin, B.T.; Dinan, T.G.; Cryan, J.F.; Hyland, N.P. Gut Reactions: Breaking Down Xenobiotic-Microbiome Interactions. Pharmacol. Rev. 2019, 71, 198–224. [Google Scholar] [CrossRef]
- Riviere, A.; Selak, M.; Lantin, D.; Leroy, F.; De Vuyst, L. Bifidobacteria and Butyrate-Producing Colon Bacteria: Importance and Strategies for Their Stimulation in the Human Gut. Front. Microbiol. 2016, 7, 979. [Google Scholar] [CrossRef]
- Guo, X.; Okpara, E.S.; Hu, W.; Yan, C.; Wang, Y.; Liang, Q.; Chiang, J.Y.L.; Han, S. Interactive Relationships between Intestinal Flora and Bile Acids. Int. J. Mol. Sci. 2022, 23, 8343. [Google Scholar] [CrossRef] [PubMed]
- Begley, M.; Hill, C.; Gahan, C.G. Bile salt hydrolase activity in probiotics. Appl. Environ. Microbiol. 2006, 72, 1729–1738. [Google Scholar] [CrossRef] [PubMed]
- Ridlon, J.M.; Harris, S.C.; Bhowmik, S.; Kang, D.J.; Hylemon, P.B. Consequences of bile salt biotransformations by intestinal bacteria. Gut. Microbes 2016, 7, 22–39. [Google Scholar] [CrossRef] [PubMed]
- Ridlon, J.M.; Daniel, S.L.; Gaskins, H.R. The Hylemon-Bjorkhem pathway of bile acid 7-dehydroxylation: History, biochemistry, and microbiology. J. Lipid Res. 2023, 64, 100392. [Google Scholar] [CrossRef] [PubMed]
- Ridlon, J.M.; Kang, D.J.; Hylemon, P.B. Bile salt biotransformations by human intestinal bacteria. J. Lipid Res. 2006, 47, 241–259. [Google Scholar] [CrossRef]
- Paik, D.; Yao, L.; Zhang, Y.; Bae, S.; D’Agostino, G.D.; Zhang, M.; Kim, E.; Franzosa, E.A.; Avila-Pacheco, J.; Bisanz, J.E.; et al. Human gut bacteria produce Tau(Eta)17-modulating bile acid metabolites. Nature 2022, 603, 907–912. [Google Scholar] [CrossRef]
- Sato, Y.; Atarashi, K.; Plichta, D.R.; Arai, Y.; Sasajima, S.; Kearney, S.M.; Suda, W.; Takeshita, K.; Sasaki, T.; Okamoto, S.; et al. Novel bile acid biosynthetic pathways are enriched in the microbiome of centenarians. Nature 2021, 599, 458–464. [Google Scholar] [CrossRef]
- Camilleri, M. Bile acid detergency: Permeability, inflammation, and effects of sulfation. Am. J. Physiol. Gastrointest. Liver Physiol. 2022, 322, G480–G488. [Google Scholar] [CrossRef]
- Sheng, W.; Ji, G.; Zhang, L. The Effect of Lithocholic Acid on the Gut-Liver Axis. Front. Pharmacol. 2022, 13, 910493. [Google Scholar] [CrossRef]
- Mousset, C.M.; Hobo, W.; Woestenenk, R.; Preijers, F.; Dolstra, H.; van der Waart, A.B. Comprehensive Phenotyping of T Cells Using Flow Cytometry. Cytom. A 2019, 95, 647–654. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Shao, Y.; Fu, R. Current research status of HLA in immune-related diseases. Immun. Inflamm. Dis. 2021, 9, 340–350. [Google Scholar] [CrossRef]
- Gomez-Bris, R.; Saez, A.; Herrero-Fernandez, B.; Rius, C.; Sanchez-Martinez, H.; Gonzalez-Granado, J.M. CD4 T-Cell Subsets and the Pathophysiology of Inflammatory Bowel Disease. Int. J. Mol. Sci. 2023, 24, 2696. [Google Scholar] [CrossRef] [PubMed]
- Kunzli, M.; Masopust, D. CD4(+) T cell memory. Nat. Immunol. 2023, 24, 903–914. [Google Scholar] [CrossRef] [PubMed]
- Knochelmann, H.M.; Dwyer, C.J.; Bailey, S.R.; Amaya, S.M.; Elston, D.M.; Mazza-McCrann, J.M.; Paulos, C.M. When worlds collide: Th17 and Treg cells in cancer and autoimmunity. Cell. Mol. Immunol. 2018, 15, 458–469. [Google Scholar] [CrossRef]
- Kiriyama, Y.; Nochi, H. Regulation of PD-L1 Expression by Nuclear Receptors. Int. J. Mol. Sci. 2023, 24, 9891. [Google Scholar] [CrossRef]
- Adams, A.B.; Ford, M.L.; Larsen, C.P. Costimulation Blockade in Autoimmunity and Transplantation: The CD28 Pathway. J. Immunol. 2016, 197, 2045–2050. [Google Scholar] [CrossRef]
- Mosmann, T.R.; Cherwinski, H.; Bond, M.W.; Giedlin, M.A.; Coffman, R.L. Two types of murine helper T cell clone. I. Definition according to profiles of lymphokine activities and secreted proteins. J. Immunol. 1986, 136, 2348–2357. [Google Scholar] [CrossRef]
- Dardalhon, V.; Awasthi, A.; Kwon, H.; Galileos, G.; Gao, W.; Sobel, R.A.; Mitsdoerffer, M.; Strom, T.B.; Elyaman, W.; Ho, I.C.; et al. IL-4 inhibits TGF-beta-induced Foxp3+ T cells and, together with TGF-beta, generates IL-9+ IL-10+ Foxp3(-) effector T cells. Nat. Immunol. 2008, 9, 1347–1355. [Google Scholar] [CrossRef]
- Veldhoen, M.; Uyttenhove, C.; van Snick, J.; Helmby, H.; Westendorf, A.; Buer, J.; Martin, B.; Wilhelm, C.; Stockinger, B. Transforming growth factor-beta ’reprograms’ the differentiation of T helper 2 cells and promotes an interleukin 9-producing subset. Nat. Immunol. 2008, 9, 1341–1346. [Google Scholar] [CrossRef]
- Langrish, C.L.; Chen, Y.; Blumenschein, W.M.; Mattson, J.; Basham, B.; Sedgwick, J.D.; McClanahan, T.; Kastelein, R.A.; Cua, D.J. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J. Exp. Med. 2005, 201, 233–240. [Google Scholar] [CrossRef] [PubMed]
- Eyerich, S.; Eyerich, K.; Pennino, D.; Carbone, T.; Nasorri, F.; Pallotta, S.; Cianfarani, F.; Odorisio, T.; Traidl-Hoffmann, C.; Behrendt, H.; et al. Th22 cells represent a distinct human T cell subset involved in epidermal immunity and remodeling. J. Clin. Investig. 2009, 119, 3573–3585. [Google Scholar] [CrossRef]
- Breitfeld, D.; Ohl, L.; Kremmer, E.; Ellwart, J.; Sallusto, F.; Lipp, M.; Forster, R. Follicular B helper T cells express CXC chemokine receptor 5, localize to B cell follicles, and support immunoglobulin production. J. Exp. Med. 2000, 192, 1545–1552. [Google Scholar] [CrossRef]
- Walker, L.S.K. The link between circulating follicular helper T cells and autoimmunity. Nat. Rev. Immunol. 2022, 22, 567–575. [Google Scholar] [CrossRef] [PubMed]
- Sakaguchi, S.; Sakaguchi, N.; Asano, M.; Itoh, M.; Toda, M. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J. Immunol. 1995, 155, 1151–1164. [Google Scholar] [CrossRef] [PubMed]
- Fontenot, J.D.; Rasmussen, J.P.; Williams, L.M.; Dooley, J.L.; Farr, A.G.; Rudensky, A.Y. Regulatory T cell lineage specification by the forkhead transcription factor foxp3. Immunity 2005, 22, 329–341. [Google Scholar] [CrossRef]
- Yu, W.; Li, C.; Zhang, D.; Li, Z.; Xia, P.; Liu, X.; Cai, X.; Yang, P.; Ling, J.; Zhang, J.; et al. Advances in T Cells Based on Inflammation in Metabolic Diseases. Cells 2022, 11, 3554. [Google Scholar] [CrossRef]
- Makishima, M.; Okamoto, A.Y.; Repa, J.J.; Tu, H.; Learned, R.M.; Luk, A.; Hull, M.V.; Lustig, K.D.; Mangelsdorf, D.J.; Shan, B. Identification of a nuclear receptor for bile acids. Science 1999, 284, 1362–1365. [Google Scholar] [CrossRef]
- Yoneno, K.; Hisamatsu, T.; Shimamura, K.; Kamada, N.; Ichikawa, R.; Kitazume, M.T.; Mori, M.; Uo, M.; Namikawa, Y.; Matsuoka, K.; et al. TGR5 signalling inhibits the production of pro-inflammatory cytokines by in vitro differentiated inflammatory and intestinal macrophages in Crohn’s disease. Immunology 2013, 139, 19–29. [Google Scholar] [CrossRef]
- He, Y.; Hara, H.; Nunez, G. Mechanism and Regulation of NLRP3 Inflammasome Activation. Trends Biochem. Sci. 2016, 41, 1012–1021. [Google Scholar] [CrossRef]
- Chen, Y.; Ye, X.; Escames, G.; Lei, W.; Zhang, X.; Li, M.; Jing, T.; Yao, Y.; Qiu, Z.; Wang, Z.; et al. The NLRP3 inflammasome: Contributions to inflammation-related diseases. Cell Mol. Biol. Lett. 2023, 28, 51. [Google Scholar] [CrossRef] [PubMed]
- Hao, H.; Cao, L.; Jiang, C.; Che, Y.; Zhang, S.; Takahashi, S.; Wang, G.; Gonzalez, F.J. Farnesoid X Receptor Regulation of the NLRP3 Inflammasome Underlies Cholestasis-Associated Sepsis. Cell Metab. 2017, 25, 856–867.e5. [Google Scholar] [CrossRef] [PubMed]
- Wammers, M.; Schupp, A.K.; Bode, J.G.; Ehlting, C.; Wolf, S.; Deenen, R.; Kohrer, K.; Haussinger, D.; Graf, D. Reprogramming of pro-inflammatory human macrophages to an anti-inflammatory phenotype by bile acids. Sci. Rep. 2018, 8, 255. [Google Scholar] [CrossRef] [PubMed]
- Shao, J.; Ge, T.; Tang, C.; Wang, G.; Pang, L.; Chen, Z. Synergistic anti-inflammatory effect of gut microbiota and lithocholic acid on liver fibrosis. Inflamm. Res. 2022, 71, 1389–1401. [Google Scholar] [CrossRef]
- Lazarevic, V.; Glimcher, L.H.; Lord, G.M. T-bet: A bridge between innate and adaptive immunity. Nat. Rev. Immunol. 2013, 13, 777–789. [Google Scholar] [CrossRef]
- Pols, T.W.H.; Puchner, T.; Korkmaz, H.I.; Vos, M.; Soeters, M.R.; de Vries, C.J.M. Lithocholic acid controls adaptive immune responses by inhibition of Th1 activation through the Vitamin D receptor. PLoS ONE 2017, 12, e0176715. [Google Scholar] [CrossRef]
- Wen, T.H.; Tsai, K.W.; Wu, Y.J.; Liao, M.T.; Lu, K.C.; Hu, W.C. The Framework for Human Host Immune Responses to Four Types of Parasitic Infections and Relevant Key JAK/STAT Signaling. Int. J. Mol. Sci. 2021, 22, 13310. [Google Scholar] [CrossRef]
- Campe, J.; Ullrich, E. T Helper Cell Lineage-Defining Transcription Factors: Potent Targets for Specific GVHD Therapy? Front. Immunol. 2021, 12, 806529. [Google Scholar] [CrossRef]
- Patel, D.D.; Kuchroo, V.K. Th17 Cell Pathway in Human Immunity: Lessons from Genetics and Therapeutic Interventions. Immunity 2015, 43, 1040–1051. [Google Scholar] [CrossRef]
- Sun, C.Y.; Yang, N.; Zheng, Z.L.; Liu, D.; Xu, Q.L. T helper 17 (Th17) cell responses to the gut microbiota in human diseases. Biomed. Pharmacother. 2023, 161, 114483. [Google Scholar] [CrossRef]
- Ivanov, I.I.; Atarashi, K.; Manel, N.; Brodie, E.L.; Shima, T.; Karaoz, U.; Wei, D.; Goldfarb, K.C.; Santee, C.A.; Lynch, S.V.; et al. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell 2009, 139, 485–498. [Google Scholar] [CrossRef] [PubMed]
- Atarashi, K.; Tanoue, T.; Ando, M.; Kamada, N.; Nagano, Y.; Narushima, S.; Suda, W.; Imaoka, A.; Setoyama, H.; Nagamori, T.; et al. Th17 Cell Induction by Adhesion of Microbes to Intestinal Epithelial Cells. Cell 2015, 163, 367–380. [Google Scholar] [CrossRef] [PubMed]
- Xiao, R.; Lei, K.; Kuok, H.; Deng, W.; Zhuang, Y.; Tang, Y.; Guo, Z.; Qin, H.; Bai, L.P.; Li, T. Synthesis and identification of lithocholic acid 3-sulfate as RORgammat ligand to inhibit Th17 cell differentiation. J. Leukoc. Biol. 2022, 112, 835–843. [Google Scholar] [CrossRef] [PubMed]
- Bluestone, J.A.; McKenzie, B.S.; Beilke, J.; Ramsdell, F. Opportunities for Treg cell therapy for the treatment of human disease. Front. Immunol. 2023, 14, 1166135. [Google Scholar] [CrossRef] [PubMed]
- Mantel, P.Y.; Ouaked, N.; Ruckert, B.; Karagiannidis, C.; Welz, R.; Blaser, K.; Schmidt-Weber, C.B. Molecular mechanisms underlying FOXP3 induction in human T cells. J. Immunol. 2006, 176, 3593–3602. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Liu, W.; Li, Y.; Liu, P.; Li, S.; Dou, D.; Wang, Y.; Yang, R.; Xiang, R.; Liu, F. Alternative Splice Variants Modulates Dominant-Negative Function of Helios in T-Cell Leukemia. PLoS ONE 2016, 11, e0163328. [Google Scholar] [CrossRef]
- Bai, L.; Hao, X.; Keith, J.; Feng, Y. DNA Methylation in Regulatory T Cell Differentiation and Function: Challenges and Opportunities. Biomolecules 2022, 12, 1282. [Google Scholar] [CrossRef]
- Lu, L.; Barbi, J.; Pan, F. The regulation of immune tolerance by FOXP3. Nat. Rev. Immunol. 2017, 17, 703–717. [Google Scholar] [CrossRef]
- Wang, K.; Fu, W. Transcriptional regulation of Treg homeostasis and functional specification. Cell. Mol. Life Sci. 2020, 77, 4269–4287. [Google Scholar] [CrossRef]
- Trujillo-Ochoa, J.L.; Kazemian, M.; Afzali, B. The role of transcription factors in shaping regulatory T cell identity. Nat. Rev. Immunol. 2023. [Google Scholar] [CrossRef]
- Sadlon, T.; Brown, C.Y.; Bandara, V.; Hope, C.M.; Schjenken, J.E.; Pederson, S.M.; Breen, J.; Forrest, A.; Beyer, M.; Robertson, S.; et al. Unravelling the molecular basis for regulatory T-cell plasticity and loss of function in disease. Clin. Transl. Immunol. 2018, 7, e1011. [Google Scholar] [CrossRef]
- Long, X.; Luo, C.; Zhu, Z. Role of CNSs Conserved Distal Cis-Regulatory Elements in CD4 + T Cell Development and Differentiation. Front. Immunol. 2022, 13, 919550. [Google Scholar] [CrossRef]
- Feng, Y.; van der Veeken, J.; Shugay, M.; Putintseva, E.V.; Osmanbeyoglu, H.U.; Dikiy, S.; Hoyos, B.E.; Moltedo, B.; Hemmers, S.; Treuting, P.; et al. A mechanism for expansion of regulatory T-cell repertoire and its role in self-tolerance. Nature 2015, 528, 132–136. [Google Scholar] [CrossRef] [PubMed]
- Nagy, G.; Nagy, L. Motif grammar: The basis of the language of gene expression. Comput. Struct. Biotechnol. J. 2020, 18, 2026–2032. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Dai, S.; Li, J.; Liang, X.; Qu, L.; Chen, X.; Guo, M.; Chen, Z.; Chen, L.; Wei, H.; et al. Structural basis of binding of homodimers of the nuclear receptor NR4A2 to selective Nur-responsive DNA elements. J. Biol. Chem. 2019, 294, 19795–19803. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Chau, T.; Liu, H.X.; Liao, D.; Keane, R.; Nie, Y.; Yang, H.; Wan, Y.J. Bile acids regulate nuclear receptor (Nur77) expression and intracellular location to control proliferation and apoptosis. Mol. Cancer Res. 2015, 13, 281–292. [Google Scholar] [CrossRef]
- Guzman, A.; Hughes, C.H.K.; Murphy, B.D. Orphan nuclear receptors in angiogenesis and follicular development. Reproduction 2021, 162, R35–R54. [Google Scholar] [CrossRef]
- Wang, T.W.; Johmura, Y.; Suzuki, N.; Omori, S.; Migita, T.; Yamaguchi, K.; Hatakeyama, S.; Yamazaki, S.; Shimizu, E.; Imoto, S.; et al. Blocking PD-L1-PD-1 improves senescence surveillance and ageing phenotypes. Nature 2022, 611, 358–364. [Google Scholar] [CrossRef]
- Yamaguchi, H.; Hsu, J.M.; Yang, W.H.; Hung, M.C. Mechanisms regulating PD-L1 expression in cancers and associated opportunities for novel small-molecule therapeutics. Nat. Rev. Clin. Oncol. 2022, 19, 287–305. [Google Scholar] [CrossRef]
- Lu, L.; Jiang, Y.X.; Liu, X.X.; Jin, J.M.; Gu, W.J.; Luan, X.; Guan, Y.Y.; Zhang, L.J. FXR agonist GW4064 enhances anti-PD-L1 immunotherapy in colorectal cancer. Oncoimmunology 2023, 12, 2217024. [Google Scholar] [CrossRef]
- Watkins, L.R.; Orlandi, C. Orphan G Protein Coupled Receptors in Affective Disorders. Genes 2020, 11, 694. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antigen-Presenting Cells | Bile Acids That Affect Antigen-Presenting Cells | Receptors for Bile Acids | Effects of Bile Acids |
|---|---|---|---|
| Dendritic cells | DCA, LCA | TGR5, FXR | Inductin of Th1 and Th17 differentiation |
| isoDCA | FXR | Inductin of Treg differentiation | |
| Macrophages | LCA, TLCA | TGR5 | Cytokine and chemokine secretion |
| T Cell Subsets | Differentiation Regulators | Characteristic Cytokines | Functions | Bile Acids That Affect T Cell Differentiation | Receptors for Bile Acids | Effects of Bile Acids |
|---|---|---|---|---|---|---|
| Th1 | T-bet, STAT1, STAT4 | IL-2, TNFα, IFNγ | cellular immunity, elimination of intracellular parasites | LCA | VDR | Th1 differentiation |
| Th17 | RORγt, STAT3 | IL-17A, IL-17F, IL-22 | host defense against extracellular pathogens, barrier protection | 3-oxoLCA, LCA-3-S | RORγt | Th17 differentiation |
| Treg | FoxP3, STAT5 | IL-10, IL-35, TGF-β | suppressing immune responses, maintaining immune tolerance | isoallo LCA | NR4A1 | Treg differentiation |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kiriyama, Y.; Nochi, H. The Role of Gut Microbiota-Derived Lithocholic Acid, Deoxycholic Acid and Their Derivatives on the Function and Differentiation of Immune Cells. Microorganisms 2023, 11, 2730. https://doi.org/10.3390/microorganisms11112730
Kiriyama Y, Nochi H. The Role of Gut Microbiota-Derived Lithocholic Acid, Deoxycholic Acid and Their Derivatives on the Function and Differentiation of Immune Cells. Microorganisms. 2023; 11(11):2730. https://doi.org/10.3390/microorganisms11112730
Chicago/Turabian StyleKiriyama, Yoshimitsu, and Hiromi Nochi. 2023. "The Role of Gut Microbiota-Derived Lithocholic Acid, Deoxycholic Acid and Their Derivatives on the Function and Differentiation of Immune Cells" Microorganisms 11, no. 11: 2730. https://doi.org/10.3390/microorganisms11112730