
Exploring the Association between Gut Microbiota and Inflammatory Skin Diseases: A Two-Sample Mendelian Randomization Analysis
 ,
,
Abstract
:1. Introduction
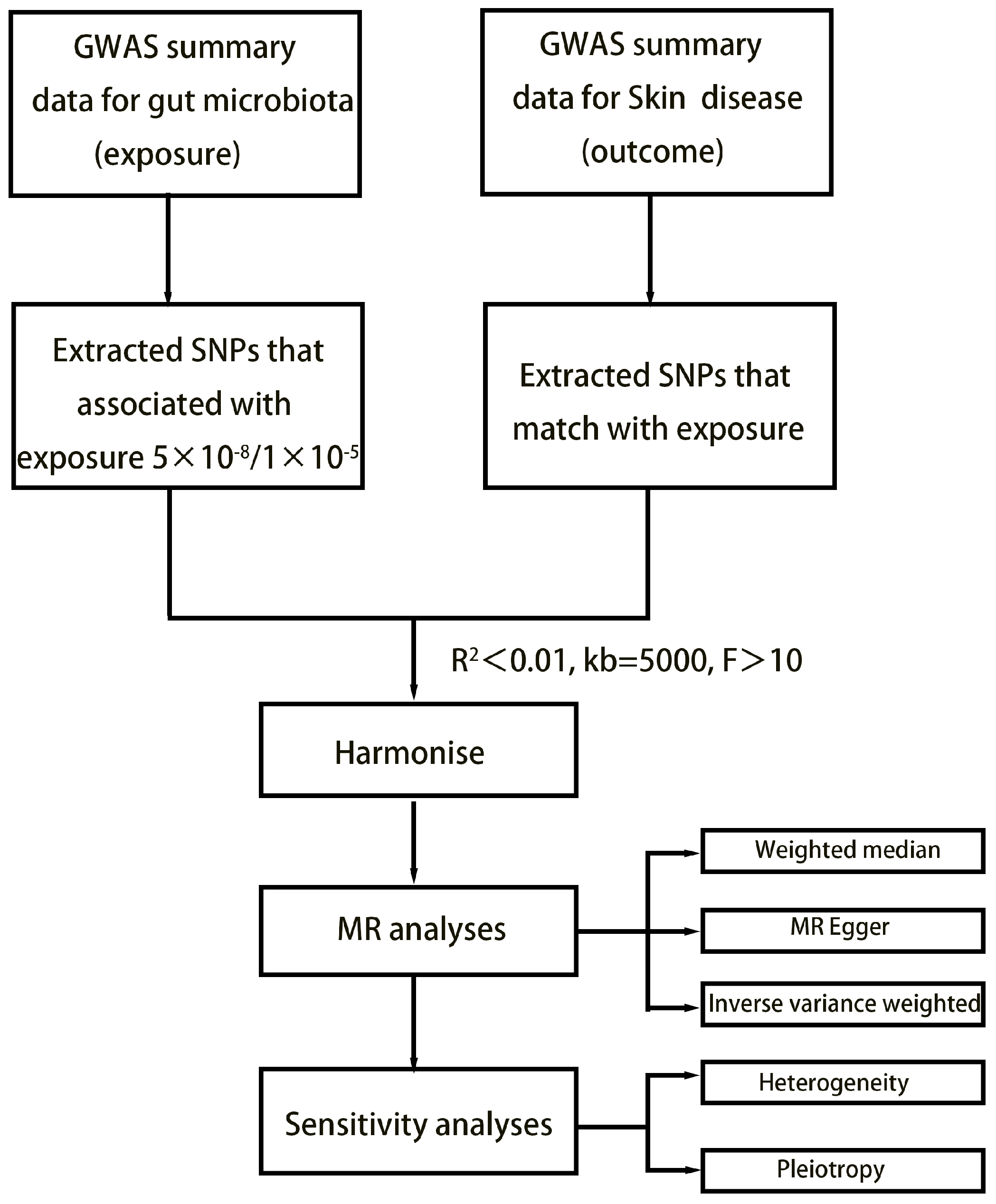
2. Method
2.1. Exposure Data
2.2. Outcome Data
2.3. Selection of Instrumental Variables
2.4. Statistical Analysis
3. Results
3.1. SNP Selection
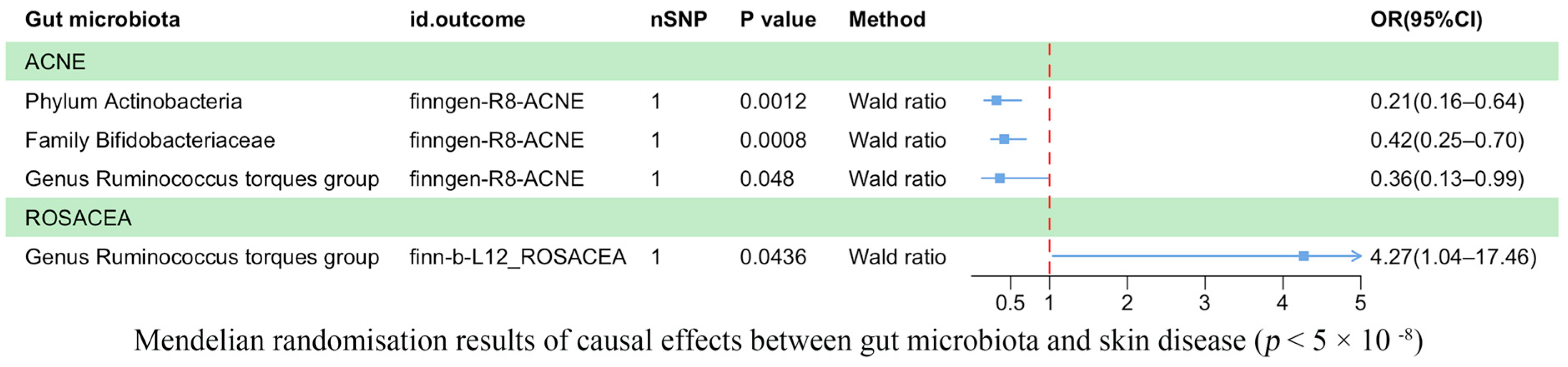
3.2. Results of the TSMR Analysis (Genome-Wide Statistical Significance Threshold, p < 5 × 10−8)
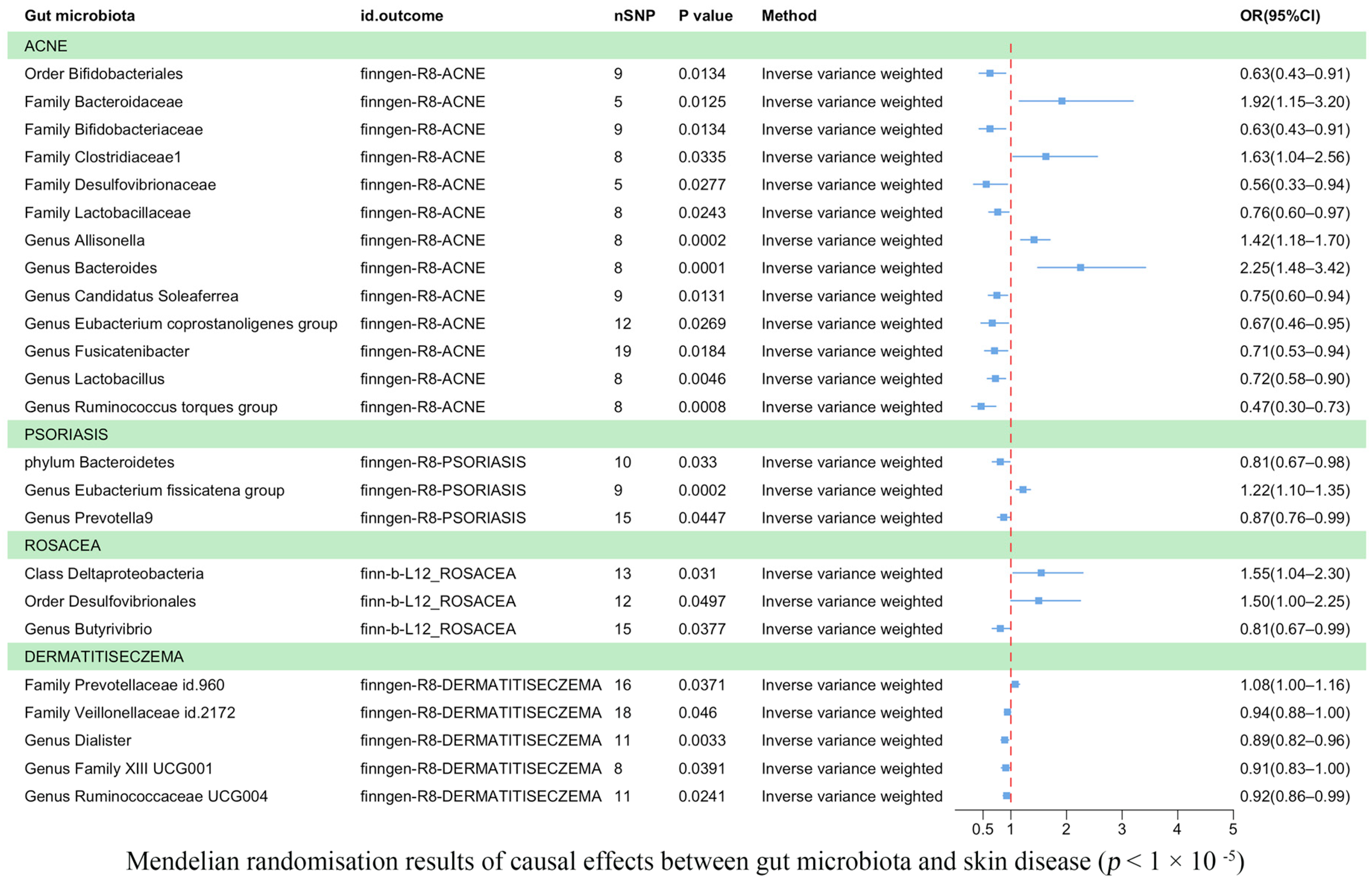
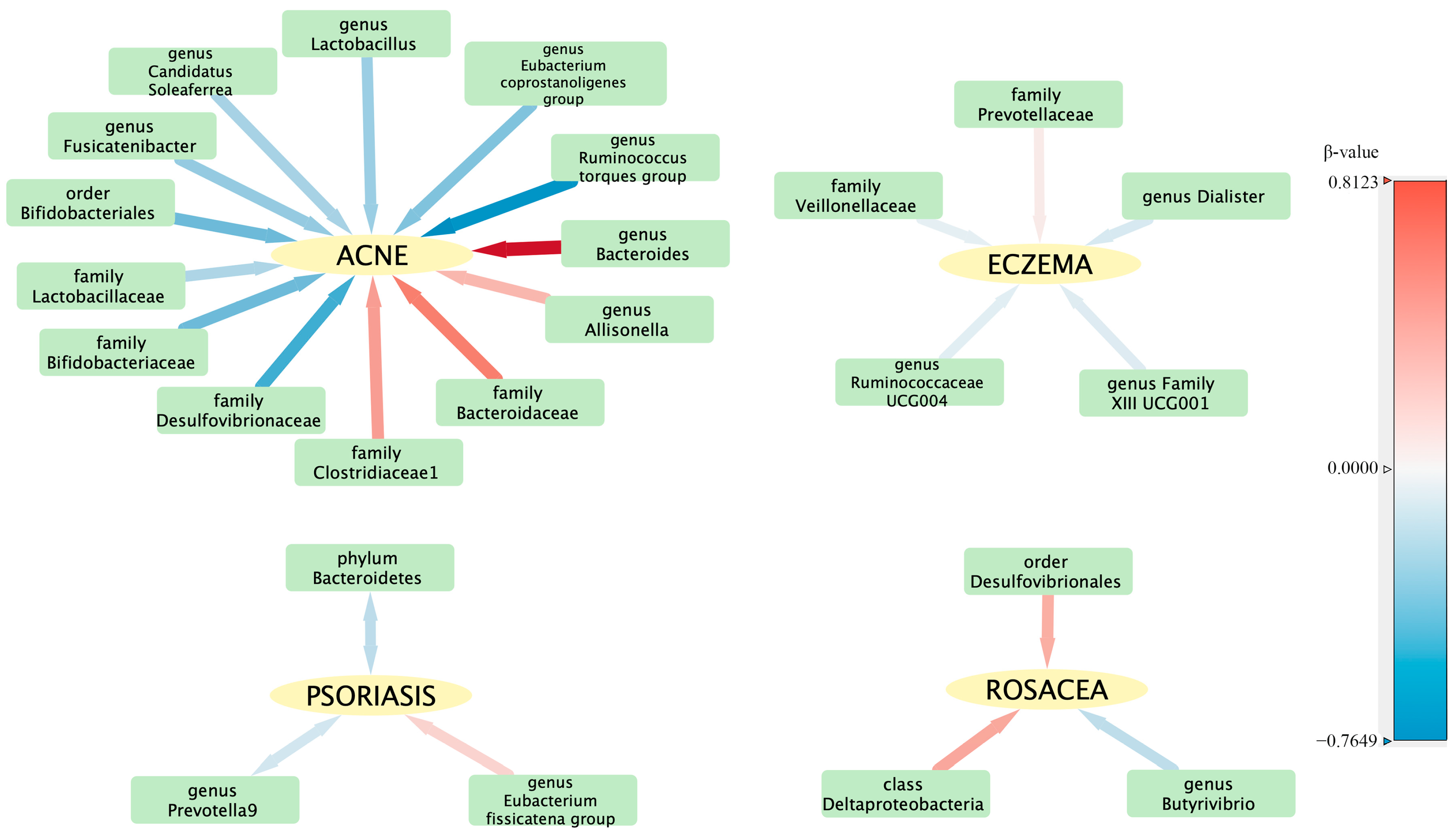
3.3. Results of the TSMR Analysis (Gene Locus Range Significance Levels, p < 1 × 10−5)
3.4. Sensitivity Analyses
3.5. Reverse TSMR Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| GWAS | Genome-wide association study |
| MR | Mendelian randomization |
| GSA | Gut–skin axis |
| SCFAs | Short-chain fatty acids |
| IVs | Instrumental variables |
| SNP | Single-nucleotide polymorphism |
| IVW | Inverse variance weighted |
References
- Karimkhani, C.; Dellavalle, R.P.; Coffeng, L.E.; Flohr, C.; Hay, R.J.; Langan, S.M.; Naghavi, M. Global Skin Disease Morbidity and Mortality: An Update from the Global Burden of Disease Study 2013. JAMA Dermatol. 2017, 153, 406–412. [Google Scholar] [CrossRef] [PubMed]
- Salem, I.; Ramser, A.; Isham, N.; Ghannoum, M.A. The Gut Microbiome as a Major Regulator of the Gut-Skin Axis. Front. Microbiol. 2018, 9, 1459. [Google Scholar] [CrossRef] [PubMed]
- Mahmud, M.R.; Akter, S.; Tamanna, S.K.; Mazumder, L.; Esti, I.Z.; Banerjee, S.; Pirttilä, A.M. Impact of gut microbiome on skin health: Gut-skin axis observed through the lenses of therapeutics and skin diseases. Gut Microbes 2022, 14, 2096995. [Google Scholar] [CrossRef]
- Willemsen, L.E.M.; Koetsier, M.A.; van Deventer, S.J.H.; Van Tol, E.A.F. Short chain fatty acids stimulate epithelial mucin 2 expression through differential effects on prostaglandin E1 and E2 production by intestinal myofibroblasts. Gut 2003, 52, 1442–1447. [Google Scholar] [CrossRef] [PubMed]
- Samuelson, D.R.; Welsh, D.A.; Shellito, J.E. Regulation of lung immunity and host defense by the intestinal microbiota. Front. Microbiol. 2015, 6, 1085. [Google Scholar] [CrossRef] [PubMed]
- Pothmann, A.; Illing, T.; Wiegand, C.; Hartmann, A.A.; Elsner, P. The Microbiome and Atopic Dermatitis: A Review. Am. J. Clin. Dermatol. 2019, 20, 749–761. [Google Scholar] [CrossRef]
- Lee, Y.B.; Byun, E.J.; Kim, H.S. Potential Role of the Microbiome in Acne: A Comprehensive Review. J. Clin. Med. 2019, 8, 987. [Google Scholar] [CrossRef]
- Yan, D.; Issa, N.; Afifi, L.; Jeon, C.; Chang, H.-W.; Liao, W. The Role of the Skin and Gut Microbiome in Psoriatic Disease. Curr. Dermatol. Rep. 2017, 6, 94–103. [Google Scholar] [CrossRef]
- Chen, Y.-J.; Lee, W.-H.; Ho, H.J.; Tseng, C.-H.; Wu, C.-Y. An altered fecal microbial profiling in rosacea patients compared to matched controls. J. Formos. Med. Assoc. 2020, 120 Pt 1, 256–264. [Google Scholar] [CrossRef]
- Dan, Y.-L.; Wang, P.; Cheng, Z.; Wu, Q.; Wang, X.-R.; Wang, D.-G.; Pan, H.-F. Circulating adiponectin levels and systemic lupus erythematosus: A two-sample Mendelian randomization study. Rheumatology 2020, 60, 940–946. [Google Scholar] [CrossRef]
- Kurilshikov, A.; Medina-Gomez, C.; Bacigalupe, R.; Radjabzadeh, D.; Wang, J.; Demirkan, A.; Le Roy, C.I.; Garay, J.A.R.; Finnicum, C.T.; Liu, X.; et al. Large-scale association analyses identify host factors influencing human gut microbiome composition. Nat. Genet. 2021, 53, 156–165. [Google Scholar] [CrossRef]
- Burgess, S.; Thompson, S.G.; CRP CHD Genetics Collaboration. Avoiding bias from weak instruments in Mendelian randomization studies. Int. J. Epidemiol. 2011, 40, 755–764. [Google Scholar] [CrossRef]
- Burgess, S.; Butterworth, A.; Thompson, S.G. Mendelian Randomization Analysis with Multiple Genetic Variants Using Summarized Data. Genet. Epidemiol. 2013, 37, 658–665. [Google Scholar] [CrossRef] [PubMed]
- Verbanck, M.; Chen, C.-Y.; Neale, B.; Do, R. Detection of widespread horizontal pleiotropy in causal relationships inferred from Mendelian randomization between complex traits and diseases. Nat. Genet. 2018, 50, 693–698. [Google Scholar] [CrossRef] [PubMed]
- Bowden, J.; Davey Smith, G.; Burgess, S. Mendelian randomization with invalid instruments: Effect estimation and bias detection through Egger regression. Int. J. Epidemiol. 2015, 44, 512–525. [Google Scholar] [CrossRef]
- Bowden, J.; Smith, G.D.; Haycock, P.C.; Burgess, S. Consistent Estimation in Mendelian Randomization with Some Invalid Instruments Using a Weighted Median Estimator. Genet. Epidemiol. 2016, 40, 304–314. [Google Scholar] [CrossRef] [PubMed]
- Burgess, S.; Dudbridge, F.; Thompson, S.G. Combining information on multiple instrumental variables in Mendelian randomization: Comparison of allele score and summarized data methods. Stat. Med. 2016, 35, 1880–1906. [Google Scholar] [CrossRef] [PubMed]
- Pagoni, P.; Dimou, N.L.; Murphy, N.; Stergiakouli, E. Using Mendelian randomisation to assess causality in observational studies. BMJ Ment. Health 2019, 22, 67–71. [Google Scholar] [CrossRef]
- Bowden, J.; Hemani, G.; Smith, G.D. Invited Commentary: Detecting Individual and Global Horizontal Pleiotropy in Mendelian Randomization—A Job for the Humble Heterogeneity Statistic? Am. J. Epidemiol. 2018, 187, 2681–2685. [Google Scholar] [CrossRef]
- Kwon, M.-S.; Lim, S.K.; Jang, J.-Y.; Lee, J.; Park, H.K.; Kim, N.; Yun, M.; Shin, M.-Y.; Jo, H.E.; Oh, Y.J.; et al. Lactobacillus sakei WIKIM30 Ameliorates Atopic Dermatitis-Like Skin Lesions by Inducing Regulatory T Cells and Altering Gut Microbiota Structure in Mice. Front. Immunol. 2018, 9, 1905. [Google Scholar] [CrossRef]
- Morotomi, M.; Nagai, F.; Sakon, H.; Tanaka, R. Dialister succinatiphilus sp. nov. and Barnesiella intestinihominis sp. nov., isolated from human faeces. Int. J. Syst. Evol. Microbiol. 2008, 58 Pt 12, 2716–2720. [Google Scholar] [CrossRef]
- Arpaia, N.; Campbell, C.; Fan, X.; Dikiy, S.; Van Der Veeken, J.; DeRoos, P.; Liu, H.; Cross, J.R.; Pfeffer, K.; Coffer, P.J.; et al. Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature 2013, 504, 451–455. [Google Scholar] [CrossRef]
- Ohira, H.; Tsutsui, W.; Fujioka, Y. Are Short Chain Fatty Acids in Gut Microbiota Defensive Players for Inflammation and Atherosclerosis? J. Atheroscler. Thromb. 2017, 24, 660–672. [Google Scholar] [CrossRef]
- Zhuge, A.; Li, B.; Yuan, Y.; Lv, L.; Li, Y.; Wu, J.; Yang, L.; Bian, X.; Wang, K.; Wang, Q.; et al. Lactobacillus salivarius LI01 encapsulated in alginate-pectin microgels ameliorates d-galactosamine-induced acute liver injury in rats. Appl. Microbiol. Biotechnol. 2020, 104, 7437–7455. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Yang, L.; Zhao, L.; Bai, F.; Liu, X. Comparison of Intestinal Microbes in Noninfectious Anterior Scleritis Patients with and Without Rheumatoid Arthritis. Front. Microbiol. 2022, 13, 925929. [Google Scholar] [CrossRef]
- Bowe, W.P.; Logan, A.C. Acne vulgaris, probiotics and the gut-brain-skin axis-back to the future? Gut Pathog. 2011, 3, 1. [Google Scholar] [CrossRef] [PubMed]
- Kurokawa, I.; Layton, A.M.; Ogawa, R. Updated Treatment for Acne: Targeted Therapy Based on Pathogenesis. Dermatol. Ther. 2021, 11, 1129–1139. [Google Scholar] [CrossRef] [PubMed]
- Salamon, M.; Sysa-Jedrzejowska, A.; Lukamowicz, J.; Lukamowicz, M.; Swiatkowska, E.; Wozniacka, A. Concentration of selected cytokines in serum of patients with acne rosacea. Przegl. Lek. 2008, 65, 371–374. [Google Scholar]
- Zafar, H.; Saier, M.H., Jr. Gut Bacteroides species in health and disease. Gut Microbes 2021, 13, 1848158. [Google Scholar] [CrossRef]
- Kim, J.M.; Cho, S.J.; Oh, Y.; Jung, H.; Kim, Y.; Kim, N. Nuclear factor-kappa B activation pathway in intestinal epithelial cells is a major regulator of chemokine gene expression and neutrophil migration induced by Bacteroides fragilis enterotoxin. Clin. Exp. Immunol. 2002, 130, 59–66. [Google Scholar] [CrossRef]
- Takeshita, K.; Mizuno, S.; Mikami, Y.; Sujino, T.; Saigusa, K.; Matsuoka, K.; Naganuma, M.; Sato, T.; Takada, T.; Tsuji, H.; et al. A Single Species of Clostridium Subcluster XIVa Decreased in Ulcerative Colitis Patients. Inflamm. Bowel Dis. 2016, 22, 2802–2810. [Google Scholar] [CrossRef]
- Caillon, F.; O’connell, M.; Eady, E.; Jenkins, G.; Cove, J.; Layton, A.; Mountford, A. Interleukin-10 secretion from CD14+ peripheral blood mononuclear cells is downregulated in patients with acne vulgaris. Br. J. Dermatol. 2010, 162, 296–303. [Google Scholar] [CrossRef] [PubMed]
- Fabbrocini, G.; Bertona, M.; Picazo, Ó.; Pareja-Galeano, H.; Monfrecola, G.; Emanuele, E. Supplementation with Lactobacillus rhamnosus SP1 normalises skin expression of genes implicated in insulin signalling and improves adult acne. Benef. Microbes 2016, 7, 625–630. [Google Scholar] [CrossRef] [PubMed]
- Sikora, M.; Stec, A.; Chrabaszcz, M.; Knot, A.; Waskiel-Burnat, A.; Rakowska, A.; Olszewska, M.; Rudnicka, L. Gut Microbiome in Psoriasis: An Updated Review. Pathogens 2020, 9, 463. [Google Scholar] [CrossRef]
- Chen, Y.; Ho, H.J.; Tseng, C.; Lai, Z.; Shieh, J.; Wu, C. Intestinal microbiota profiling and predicted metabolic dysregulation in psoriasis patients. Exp. Dermatol. 2018, 27, 1336–1343. [Google Scholar] [CrossRef]
- Kim, J.; Moreno, A.; Krueger, J.G. The imbalance between Type 17 T-cells and regulatory immune cell subsets in psoriasis vulgaris. Front. Immunol. 2022, 13, 1005115. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Liu, Q.; Wu, H.; Tang, T.; Zhao, T.; Li, Z. The dysbiosis gut microbiota induces the alternation of metabolism and imbalance of Th17/Treg in OSA patients. Arch. Microbiol. 2022, 204, 217. [Google Scholar]
- Louis, P.; Scott, K.P.; Duncan, S.H.; Flint, H.J. Understanding the effects of diet on bacterial metabolism in the large intestine. J. Appl. Microbiol. 2007, 102, 1197–1208. [Google Scholar] [CrossRef]
- Chang, P.V.; Hao, L.; Offermanns, S.; Medzhitov, R. The microbial metabolite butyrate regulates intestinal macrophage function via histone deacetylase inhibition. Proc. Natl. Acad. Sci. USA 2014, 111, 2247–2252. [Google Scholar] [CrossRef]
- Schwarz, A.; Philippsen, R.; Schwarz, T. Induction of Regulatory T Cells and Correction of Cytokine Disbalance by Short-Chain Fatty Acids: Implications for Psoriasis Therapy. J. Investig. Dermatol. 2020, 141, 95–104.e2. [Google Scholar] [CrossRef]
- Salonen, A.; Lahti, L.; Salojärvi, J.; Holtrop, G.; Korpela, K.; Duncan, S.H.; Date, P.; Farquharson, F.; Johnstone, A.M.; Lobley, G.E.; et al. Impact of diet and individual variation on intestinal microbiota composition and fermentation products in obese men. ISME J. 2014, 8, 2218–2230. [Google Scholar] [CrossRef] [PubMed]
- Chiodini, R.J.; Dowd, S.E.; Chamberlin, W.M.; Galandiuk, S.; Davis, B.; Glassing, A. Microbial Population Differentials between Mucosal and Submucosal Intestinal Tissues in Advanced Crohn’s Disease of the Ileum. PLoS ONE 2015, 10, e0134382. [Google Scholar] [CrossRef] [PubMed]
- Holmes, A.D.; Spoendlin, J.; Chien, A.L.; Baldwin, H.; Chang, A.L.S. Evidence-based update on rosacea comorbidities and their common physiologic pathways. J. Am. Acad. Dermatol. 2018, 78, 156–166. [Google Scholar] [CrossRef] [PubMed]
- Zhen, Z.; Xia, L.; You, H.; Jingwei, Z.; Shasha, Y.; Xinyi, W.; Wenjing, L.; Xin, Z.; Chaomei, F. An Integrated Gut Microbiota and Network Pharmacology Study on Fuzi-Lizhong Pill for Treating Diarrhea-Predominant Irritable Bowel Syndrome. Front. Pharmacol. 2021, 12, 746923. [Google Scholar] [CrossRef] [PubMed]
- Holmes, A.D.; Steinhoff, M. Integrative concepts of rosacea pathophysiology, clinical presentation and new therapeutics. Exp. Dermatol. 2016, 26, 659–667. [Google Scholar] [CrossRef]
- Saber, W.I.A.; Ghoniem, A.A.; Al-Otibi, F.O.; El-Hersh, M.S.; Eldadamony, N.M.; Menaa, F.; Elattar, K.M. A comparative study using response surface methodology and artificial neural network towards optimized production of melanin by Aureobasidium pullulans AKW. Sci. Rep. 2023, 13, 13545. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Threshold/Method | Outcome | Id. Exposure | Id. Outcome | Nsnp | Beta | Se | p-Value | Trait |
|---|---|---|---|---|---|---|---|---|
| 5 × 10−8/Wald ratio | ACNE | ebi-a-GCST90017110 | finngen-R8-ACNE | 1 | −1.14272 | 0.351571 | 0.001153 | Phylum Actinobacteria id.400 |
| 1 × 10−5/IVW | ACNE | ebi-a-GCST90017093 | finngen-R8-ACNE | 9 | −0.46978 | 0.190061 | 0.013446 | Order Bifidobacteriales id.432 |
| 5 × 10−8/Wald ratio | ACNE | ebi-a-GCST90016929 | finngen-R8-ACNE | 1 | −0.875 | 0.26169 | 0.000827 | Family Bifidobacteriaceae id.433 |
| 1 × 10−5/IVW | ACNE | ebi-a-GCST90016927 | finngen-R8-ACNE | 5 | 0.651397 | 0.260806 | 0.012503 | Family Bacteroidaceae id.917 |
| 1 × 10−5/IVW | ACNE | ebi-a-GCST90016929 | finngen-R8-ACNE | 9 | −0.46978 | 0.190061 | 0.013446 | Family Bifidobacteriaceae id.433 |
| 1 × 10−5/IVW | ACNE | ebi-a-GCST90016931 | finngen-R8-ACNE | 8 | 0.488064 | 0.229627 | 0.033548 | Family Clostridiaceae1 id.1869 |
| 1 × 10−5/IVW | ACNE | ebi-a-GCST90016935 | finngen-R8-ACNE | 5 | −0.58438 | 0.265392 | 0.027668 | Family Desulfovibrionaceae id.3169 |
| 1 × 10−5/IVW | ACNE | ebi-a-GCST90016941 | finngen-R8-ACNE | 8 | −0.26929 | 0.119557 | 0.024295 | Family Lactobacillaceae id.1836 |
| 5 × 10−8/Wald ratio | ACNE | ebi-a-GCST90017066 | finngen-R8-ACNE | 1 | −1.0184 | 0.515063 | 0.048014 | Genus Ruminococcus torques group id.14377 |
| 1 × 10−5/IVW | ACNE | ebi-a-GCST90016963 | finngen-R8-ACNE | 8 | 0.347787 | 0.094021 | 0.000216 | Genus Allisonella id.2174 |
| 1 × 10−5/IVW | ACNE | ebi-a-GCST90016968 | finngen-R8-ACNE | 8 | 0.812298 | 0.213034 | 0.000137 | Genus Bacteroides id.918 |
| 1 × 10−5/IVW | ACNE | ebi-a-GCST90016976 | finngen-R8-ACNE | 9 | −0.28914 | 0.116587 | 0.013137 | Genus Candidatus Soleaferrea id.11350 |
| 1 × 10−5/IVW | ACNE | ebi-a-GCST90016997 | finngen-R8-ACNE | 12 | −0.40734 | 0.184114 | 0.026938 | Genus Eubacterium coprostanoligenes group id.11375 |
| 1 × 10−5/IVW | ACNE | ebi-a-GCST90017011 | finngen-R8-ACNE | 19 | −0.34742 | 0.147413 | 0.018436 | Genus Fusicatenibacter id.11305 |
| 1 × 10−5/IVW | ACNE | ebi-a-GCST90017030 | finngen-R8-ACNE | 8 | −0.32463 | 0.11442 | 0.004552 | Genus Lactobacillus id.1837 |
| 1 × 10−5/IVW | ACNE | ebi-a-GCST90017066 | finngen-R8-ACNE | 8 | −0.76493 | 0.229111 | 0.000842 | Genus Ruminococcus torques group id.14377 |
| 1 × 10−5/IVW | PSORIASIS | ebi-a-GCST90017111 | finngen-R8-PSORIASIS | 10 | −0.21072 | 0.098831 | 0.032997 | Phylum Bacteroidetes id.905 |
| 1 × 10−5/IVW | PSORIASIS | ebi-a-GCST90016999 | finngen-R8-PSORIASIS | 9 | 0.197321 | 0.052692 | 0.000181 | Genus Eubacterium fissicatena group id.14373 |
| 1 × 10−5/IVW | PSORIASIS | ebi-a-GCST90017045 | finngen-R8-PSORIASIS | 15 | −0.13764 | 0.068571 | 0.044716 | Genus Prevotella 9 id.11183 |
| 1 × 10−5/IVW | ROSACEA | ebi-a-GCST90016915 | finn-b-L12_ROSACEA | 13 | 0.435394 | 0.201843 | 0.030998 | Class Deltaproteobacteria id.3087 |
| 1 × 10−5/IVW | ROSACEA | ebi-a-GCST90017097 | finn-b-L12_ROSACEA | 12 | 0.405676 | 0.2067 | 0.049689 | Order Desulfovibrionales id.3156 |
| 5 × 10−8/Wald ratio | ROSACEA | ebi-a-GCST90017066 | finn-b-L12_ROSACEA | 1 | 1.450923 | 0.718926 | 0.043572 | Genus Ruminococcus torques group id.14377 |
| 1 × 10−5/IVW | ROSACEA | ebi-a-GCST90016975 | finn-b-L12_ROSACEA | 15 | −0.20966 | 0.100904 | 0.037723 | Genus Butyrivibrio id.1993 |
| 1 × 10−5/IVW | DERMATITISECZEMA | ebi-a-GCST90016948 | finngen-R8-DERMATITISECZEMA | 16 | 0.074607 | 0.035786 | 0.037084 | Family Prevotellaceae id.960 |
| 1 × 10−5/IVW | DERMATITISECZEMA | ebi-a-GCST90016956 | finngen-R8-DERMATITISECZEMA | 18 | −0.06238 | 0.031264 | 0.046024 | Family Veillonellaceae id.2172 |
| 1 × 10−5/IVW | DERMATITISECZEMA | ebi-a-GCST90016988 | finngen-R8-DERMATITISECZEMA | 11 | −0.11711 | 0.039833 | 0.003282 | Genus Dialister id.2183 |
| 1 × 10−5/IVW | DERMATITISECZEMA | ebi-a-GCST90017009 | finngen-R8-DERMATITISECZEMA | 8 | −0.09576 | 0.046417 | 0.039115 | Genus Family XIII UCG001 id.11294 |
| 1 × 10−5/IVW | DERMATITISECZEMA | ebi-a-GCST90017055 | finngen-R8-DERMATITISECZEMA | 11 | −0.07922 | 0.035124 | 0.024099 | Genus Ruminococcaceae UCG004 id.11362 |
| Id. Outcome | Gut Microbiota (Outcome) | Exposure | Method | Number of Snps | Beta | Se | p-Value | Correct_Causal Direction | Steiger_Pval |
|---|---|---|---|---|---|---|---|---|---|
| ebi-a-GCST90017045 | genus Prevotella 9 | PSORIASIS | IVW | 75 | 0.04 | 0.02 | 0.02 | TRUE | 0.001 |
| ebi-a-GCST90017111 | phylum Bacteroidetes | PSORIASIS | IVW | 80 | 0.04 | 0.01 | 0.002 | TRUE | 0.0002 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Long, J.; Gu, J.; Yang, J.; Chen, P.; Dai, Y.; Lin, Y.; Wu, M.; Wu, Y. Exploring the Association between Gut Microbiota and Inflammatory Skin Diseases: A Two-Sample Mendelian Randomization Analysis. Microorganisms 2023, 11, 2586. https://doi.org/10.3390/microorganisms11102586
Long J, Gu J, Yang J, Chen P, Dai Y, Lin Y, Wu M, Wu Y. Exploring the Association between Gut Microbiota and Inflammatory Skin Diseases: A Two-Sample Mendelian Randomization Analysis. Microorganisms. 2023; 11(10):2586. https://doi.org/10.3390/microorganisms11102586
Chicago/Turabian StyleLong, Junhao, Jinglan Gu, Juexi Yang, Pu Chen, Yan Dai, Yun Lin, Ming Wu, and Yan Wu. 2023. "Exploring the Association between Gut Microbiota and Inflammatory Skin Diseases: A Two-Sample Mendelian Randomization Analysis" Microorganisms 11, no. 10: 2586. https://doi.org/10.3390/microorganisms11102586