

Abnormal Development of Microbiota May Be a Risk Factor for Febrile Urinary Tract Infection in Infancy
 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Participants
2.2. Sampling and Measurement
2.3. Statistical Analysis
2.4. Institutional Review Board Statement
3. Results
3.1. Profiles of Study Participants
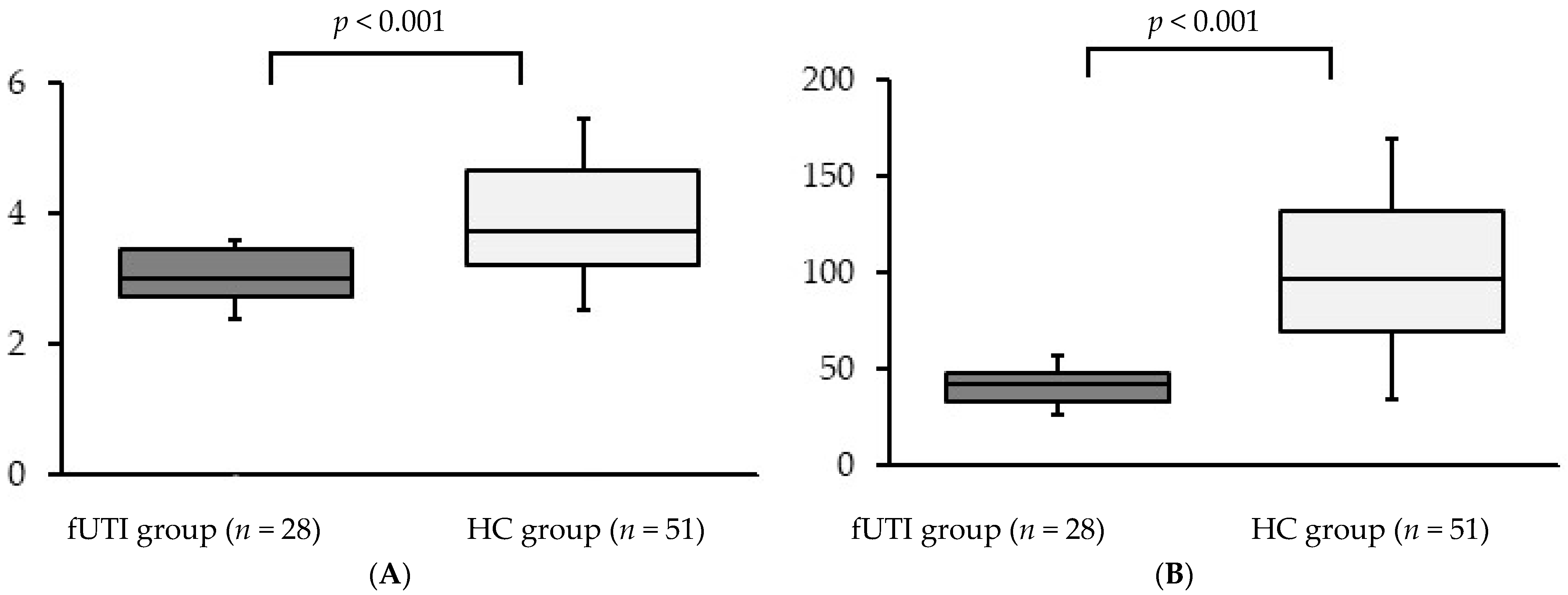
3.2. Alpha Diversity
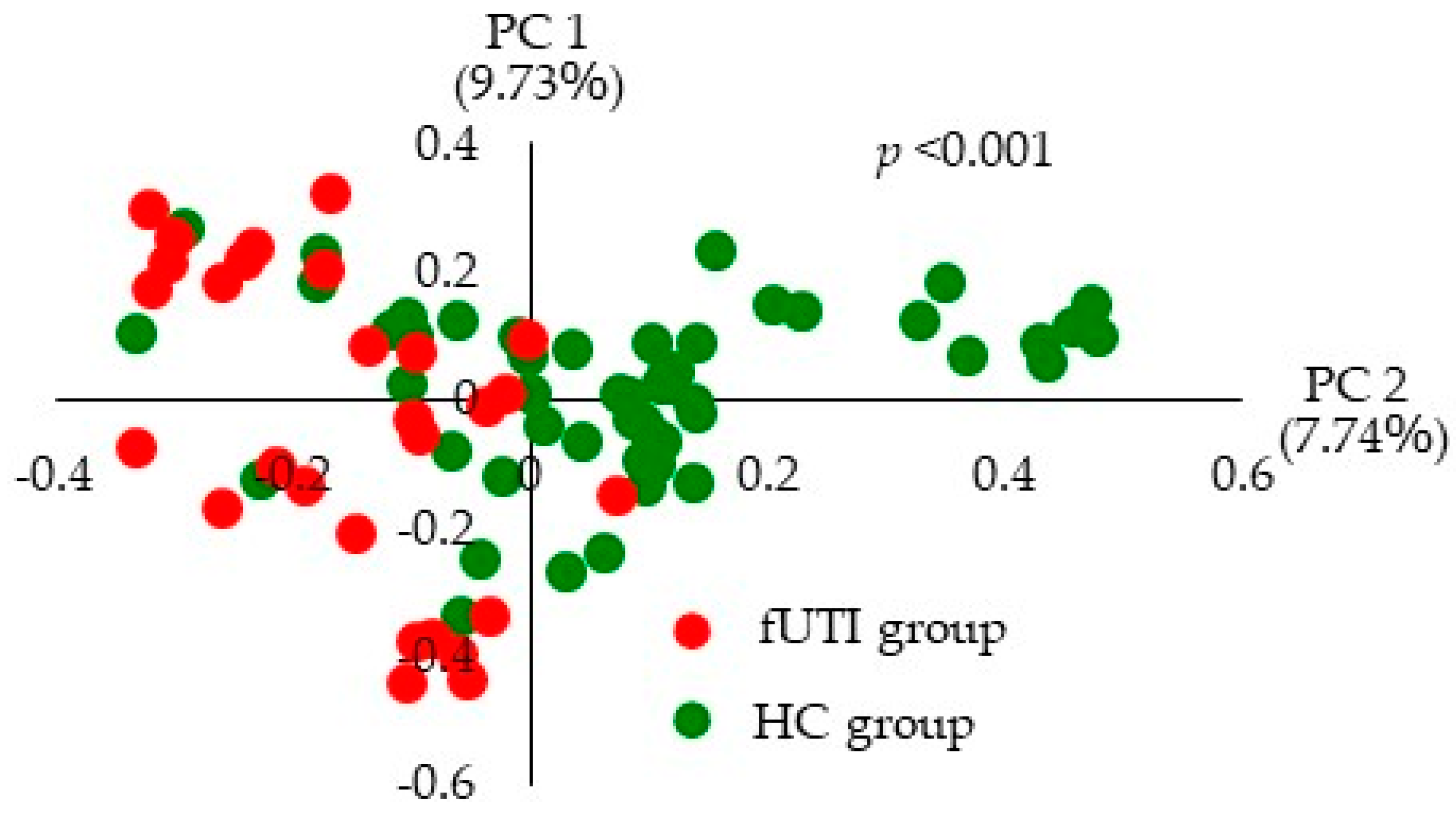
3.3. Beta Diversity
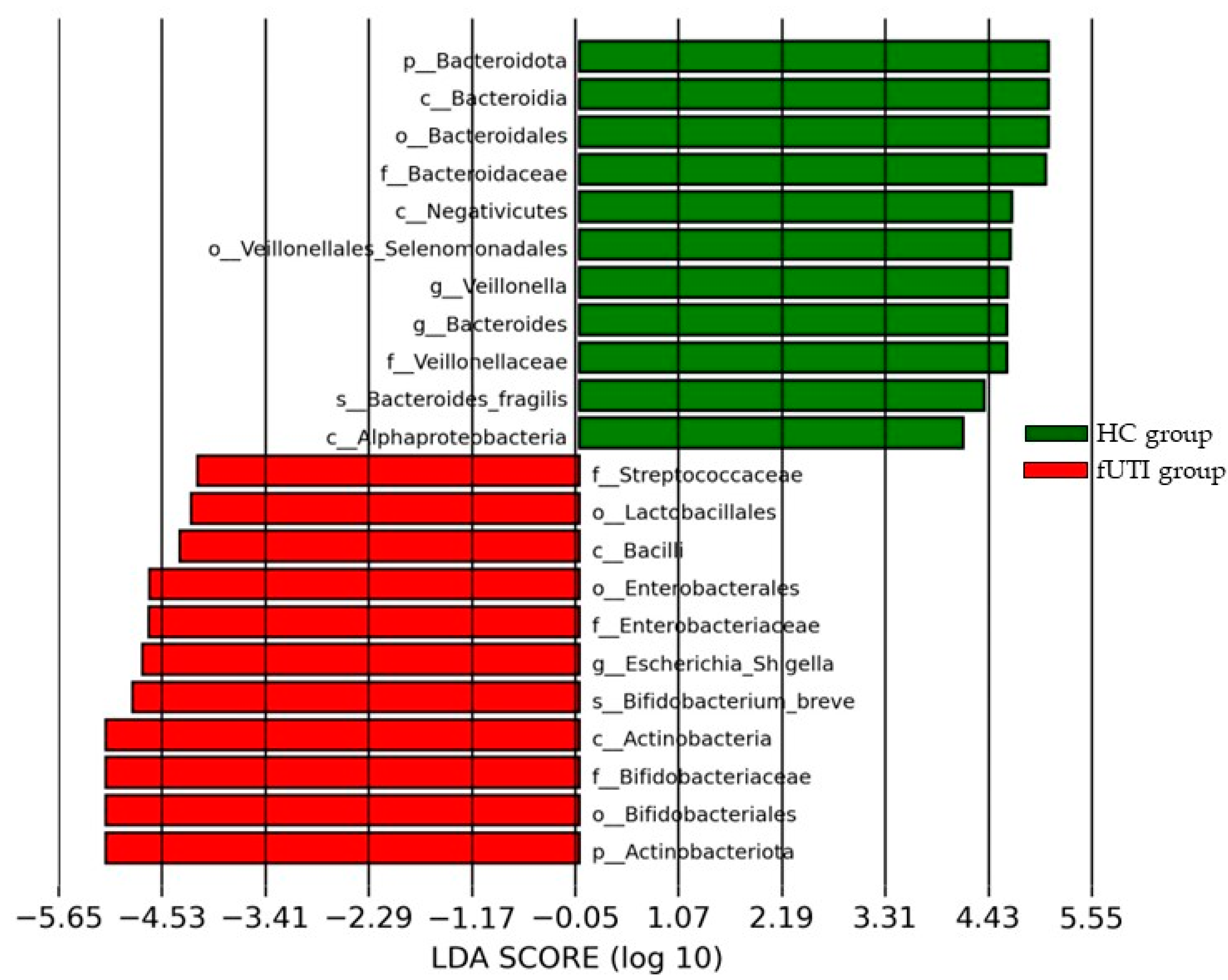
3.4. LEfSe Analysis
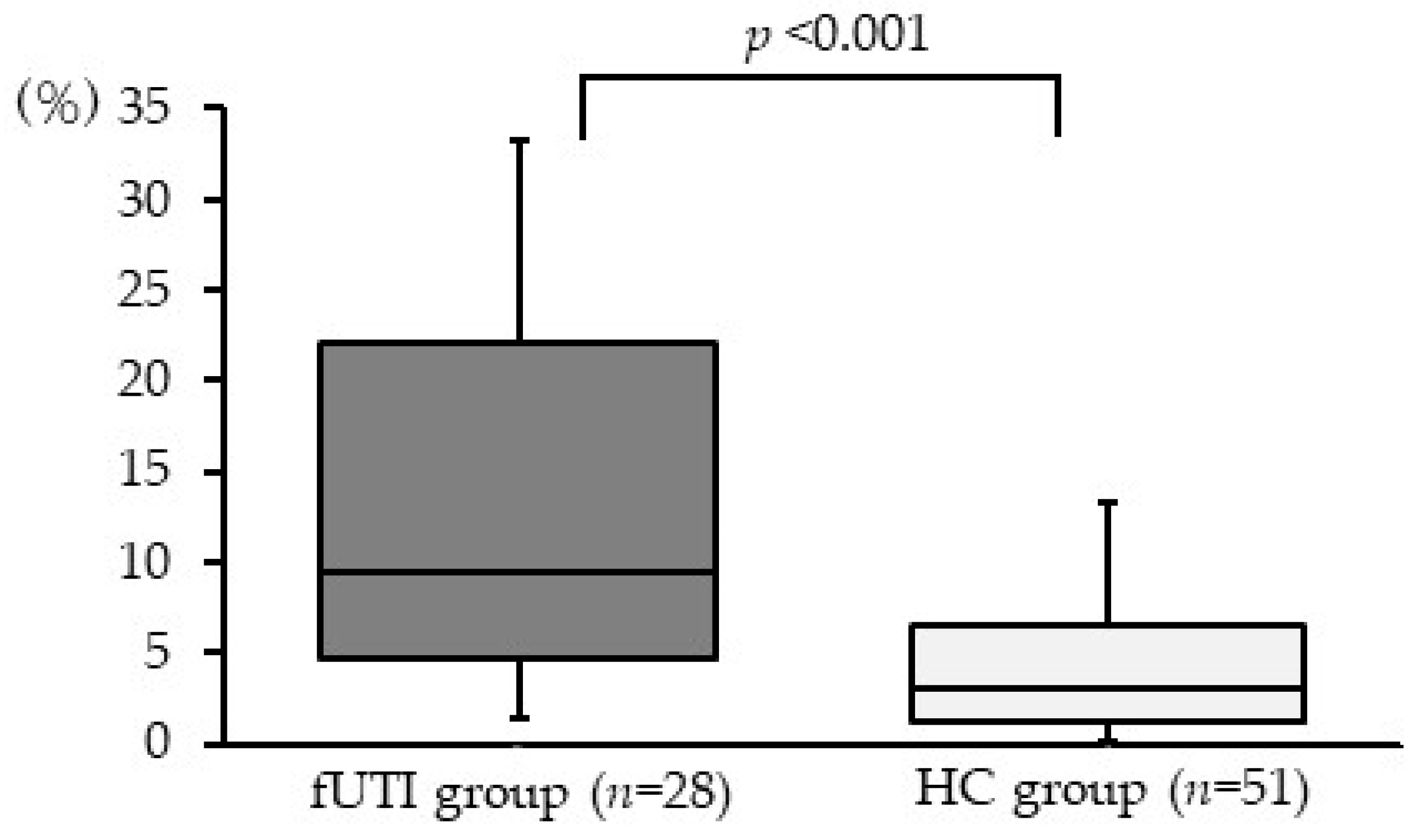
3.5. Relative Abundance of Genus Escherichia-Shigella in Gut Microbiota
4. Discussion
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hellström, A.; Hanson, E.; Hansson, S.; Hjälmås, K.; Jodal, U. Association between urinary symptoms at 7 years old and previous urinary tract infection. Arch. Dis. Child. 1991, 66, 232–234. [Google Scholar] [CrossRef]
- Freedman, A.L.; Urologic Diseases in America Project. Urologic diseases in North America Project: Trends in resource utilization for urinary tract infections in children. J. Urol. 2005, 173, 949–954. [Google Scholar] [CrossRef]
- Shaikh, N.; Morone, N.E.; Bost, J.E.; Farrell, M.H. Prevalence of urinary tract infection in childhood: A meta-analysis. Pediatr. Infect. Dis. J. 2008, 27, 302–308. [Google Scholar] [CrossRef] [PubMed]
- Stein, R.; Dogan, H.S.; Hoebeke, P.; Kočvara, R.; Nijman, R.J.; Radmayr, C.; Tekgül, S.; European Association of Urology; European Society for Pediatric Urology. Urinary tract infections in children: EAU/ESPU guidelines. Eur. Urol. 2015, 67, 546–558. [Google Scholar] [CrossRef] [PubMed]
- Jodal, U. The natural history of bacteriuria in childhood. Infect. Dis. Clin. N. Am. 1987, 1, 713–729. [Google Scholar] [CrossRef]
- Shaikh, N.; Craig, J.C.; Rovers, M.M.; Da Dalt, L.; Gardikis, S.; Hoberman, A.; Montini, G.; Rodrigo, C.; Taskinen, S.; Tuerlinckx, D.; et al. Identification of children and adolescents at risk for renal scarring after a first urinary tract infection: A meta-analysis with individual patient data. JAMA Pediatr. 2014, 168, 893–900. [Google Scholar] [CrossRef]
- Shaikh, N.; Haralam, M.A.; Kurs-Lasky, M.; Hoberman, A. Association of Renal Scarring with Number of Febrile Urinary Tract Infections in Children. JAMA Pediatr. 2019, 173, 949–952. [Google Scholar] [CrossRef] [PubMed]
- Zareba, P.; Lorenzo, A.J.; Braga, L.H. Risk factors for febrile urinary tract infection in infants with prenatal hydronephrosis: Comprehensive single center analysis. J. Urol. 2014, 191, 1614–1618. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Zhou, J.; Wang, L. Role and Mechanism of Gut Microbiota in Human Disease. Front. Cell. Infect. Microbiol. 2021, 11, 625913. [Google Scholar] [CrossRef]
- Ihekweazu, F.D.; Versalovic, J. Development of the Pediatric Gut Microbiome: Impact on Health and Disease. Am. J. Med. Sci. 2018, 356, 413–423. [Google Scholar] [CrossRef]
- Akagawa, S.; Akagawa, Y.; Yamanouchi, S.; Kimata, T.; Tsuji, S.; Kaneko, K. Development of the gut microbiota and dysbiosis in children. Biosci. Microbiota Food Health 2021, 40, 12–18. [Google Scholar] [CrossRef] [PubMed]
- Tsuji, S.; Suruda, C.; Hashiyada, M.; Kimata, T.; Yamanouchi, S.; Kitao, T.; Kino, J.; Akane, A.; Kaneko, K. Gut Microbiota Dysbiosis in Children with Relapsing Idiopathic Nephrotic Syndrome. Am. J. Nephrol. 2018, 47, 164–170. [Google Scholar] [CrossRef] [PubMed]
- Yamagishi, M.; Akagawa, S.; Akagawa, Y.; Nakai, Y.; Yamanouchi, S.; Kimata, T.; Hashiyada, M.; Akane, A.; Tsuji, S.; Kaneko, K. Decreased butyric acid-producing bacteria in gut microbiota of children with egg allergy. Allergy 2021, 76, 2279–2282. [Google Scholar] [CrossRef]
- Fujishiro, S.; Tsuji, S.; Akagawa, S.; Akagawa, Y.; Yamanouchi, S.; Ishizaki, Y.; Hashiyada, M.; Akane, A.; Kaneko, K. Dysbiosis in Gut Microbiota in Children Born Preterm Who Developed Autism Spectrum Disorder: A Pilot Study. J. Autism. Dev. Disord. 2023, 53, 4012–4020. [Google Scholar] [CrossRef] [PubMed]
- Lutter, S.A.; Currie, M.L.; Mitz, L.B.; Greenbaum, L.A. Antibiotic resistance patterns in children hospitalized for urinary tract infections. Arch. Pediatr. Adolesc. Med. 2005, 159, 924–928. [Google Scholar] [CrossRef]
- Chakupurakal, R.; Ahmed, M.; Sobithadevi, D.N.; Chinnappan, S.; Reynolds, T. Urinary tract pathogens and resistance pattern. J. Clin. Pathol. 2010, 63, 652–654. [Google Scholar] [CrossRef]
- Suh, W.; Kim, B.N.; Kang, H.M.; Yang, E.A.; Rhim, J.W.; Lee, K.Y. Febrile urinary tract infection in children: Changes in epidemiology, etiology, and antibiotic resistance patterns over a decade. Clin. Exp. Pediatr. 2021, 64, 293–300. [Google Scholar] [CrossRef]
- Alberici, I.; Bayazit, A.K.; Drozdz, D.; Emre, S.; Fischbach, M.; Harambat, J.; Jankauskiene, A.; Litwin, M.; Mir, S.; Morello, W.; et al. Pathogens causing urinary tract infections in infants: A European overview by the ESCAPE study group. Eur. J. Pediatr. 2015, 174, 783–790. [Google Scholar] [CrossRef] [PubMed]
- Paalanne, N.; Husso, A.; Salo, J.; Pieviläinen, O.; Tejesvi, M.V.; Koivusaari, P.; Pirttilä, A.M.; Pokka, T.; Mattila, S.; Jyrkäs, J.; et al. Intestinal microbiome as a risk factor for urinary tract infections in children. Eur. J. Clin. Microbiol. Infect. Dis. 2018, 37, 1881–1891. [Google Scholar] [CrossRef]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef] [PubMed]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [PubMed]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic. Acids. Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef] [PubMed]
- Lozupone, C.; Lladser, M.E.; Knights, D.; Stombaugh, J.; Knight, R. UniFrac: An effective distance metric for microbial community comparison. ISME J. 2011, 5, 169–172. [Google Scholar] [CrossRef] [PubMed]
- Faul, F.; Erdfelder, E.; Lang, A.G.; Buchner, A. G*Power 3: A flexible statistical power analysis program for the social, behavioral, and biomedical sciences. Behav. Res. Methods. 2007, 39, 175–191. [Google Scholar] [CrossRef]
- Magruder, M.; Edusei, E.; Zhang, L.; Albakry, S.; Satlin, M.J.; Westblade, L.F.; Malha, L.; Sze, C.; Lubetzky, M.; Dadhania, D.M.; et al. Gut commensal microbiota and decreased risk for Enterobacteriaceae bacteriuria and urinary tract infection. Gut. Microbes. 2020, 12, 1805281. [Google Scholar] [CrossRef]
- Magruder, M.; Sholi, A.N.; Gong, C.; Zhang, L.; Edusei, E.; Huang, J.; Albakry, S.; Satlin, M.J.; Westblade, L.F.; Crawford, C.; et al. Gut uropathogen abundance is a risk factor for development of bacteriuria and urinary tract infection. Nat. Commun. 2019, 10, 5521. [Google Scholar] [CrossRef]
- Kriss, M.; Hazleton, K.Z.; Nusbacher, N.M.; Martin, C.G.; Lozupone, C.A. Low diversity gut microbiota dysbiosis: Drivers, functional implications and recovery. Curr. Opin. Microbiol. 2018, 44, 34–40. [Google Scholar] [CrossRef]
- Eribo, O.A.; du Plessis, N.; Chegou, N.N. The Intestinal Commensal, Bacteroides fragilis, Modulates Host Responses to Viral Infection and Therapy: Lessons for Exploration during Mycobacterium tuberculosis Infection. Infect. Immun. 2022, 90, e0032121. [Google Scholar] [CrossRef]
- Erturk-Hasdemir, D.; Ochoa-Repáraz, J.; Kasper, D.L.; Kasper, L.H. Exploring the Gut-Brain Axis for the Control of CNS Inflammatory Demyelination: Immunomodulation by Bacteroides fragilis’ Polysaccharide A. Front. Immunol. 2021, 12, 662807. [Google Scholar] [CrossRef]
- Chichlowski, M.; Shah, N.; Wampler, J.L.; Wu, S.S.; Vanderhoof, J.A. Bifidobacterium longum Subspecies infantis (B. infantis) in Pediatric Nutrition: Current State of Knowledge. Nutrients 2020, 12, 1581. [Google Scholar] [CrossRef]
- Shao, T.; Hsu, R.; Hacein-Bey, C.; Zhang, W.; Gao, L.; Kurth, M.J.; Zhao, H.; Shuai, Z.; Leung, P.S.C. The Evolving Landscape of Fecal Microbial Transplantation. Clin. Rev. Allergy Immunol. 2023, 9, 101–120. [Google Scholar] [CrossRef] [PubMed]
- Hamamah, S.; Gheorghita, R.; Lobiuc, A.; Sirbu, I.O.; Covasa, M. Fecal microbiota transplantation in non-communicable diseases: Recent advances and protocols. Front. Med. 2022, 9, 1060581. [Google Scholar] [CrossRef]
- Bier, N.; Hanson, B.; Jiang, Z.D.; DuPont, H.L.; Arias, C.A.; Miller, W.R. A Case of Successful Treatment of Recurrent Urinary Tract Infection by Extended-Spectrum β-Lactamase Producing Klebsiella pneumoniae Using Oral Lyophilized Fecal Microbiota Transplant. Microb. Drug. Resist. 2023, 29, 34–38. [Google Scholar] [CrossRef] [PubMed]
- Aira, A.; Rubio, E.; Vergara Gómez, A.; Fehér, C.; Casals-Pascual, C.; González, B.; Morata, L.; Rico, V.; Soriano, A. rUTI Resolution After FMT for Clostridioides difficile Infection: A Case Report. Infect. Dis. Ther. 2021, 10, 1065–1071. [Google Scholar] [CrossRef] [PubMed]
- Jeney, S.E.S.; Lane, F.; Oliver, A.; Whiteson, K.; Dutta, S. Fecal Microbiota Transplantation for the Treatment of Refractory Recurrent Urinary Tract Infection. Obstet. Gynecol. 2020, 136, 771–773. [Google Scholar] [CrossRef] [PubMed]
- Biehl, L.M.; Cruz Aguilar, R.; Farowski, F.; Hahn, W.; Nowag, A.; Wisplinghoff, H.; Vehreschild, M.J.G.T. Fecal microbiota transplantation in a kidney transplant recipient with recurrent urinary tract infection. Infection 2018, 46, 871–874. [Google Scholar] [CrossRef]
- Jeong, J.; Yun, K.; Mun, S.; Chung, W.H.; Choi, S.Y.; Nam, Y.D.; Lim, M.Y.; Hong, C.P.; Park, C.; Ahn, Y.J.; et al. The effect of taxonomic classification by full-length 16S rRNA sequencing with a synthetic long-read technology. Sci. Rep. 2021, 11, 1727. [Google Scholar] [CrossRef]
- Yamaguchi, T.; Tsuji, S.; Akagawa, S.; Akagawa, Y.; Kino, J.; Yamanouchi, S.; Kimata, T.; Hashiyada, M.; Akane, A.; Kaneko, K. Clinical Significance of Probiotics for Children with Idiopathic Nephrotic Syndrome. Nutrients 2021, 13, 365. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| fUTI Group (n = 28) | HC Group (n = 51) | p Value | |
|---|---|---|---|
| Age, months (median, IQR) | 5 (3.8–7.0) | 5 (4.0–8.0) | 0.40 |
| Sex (Boy: Girl) | 14:14 | 28:23 | 0.86 |
| Gestational age, weeks (median, IQR) | 39.2 (38.1–40.1) | 39.6 (38.4–40.4) | 0.20 |
| Mode of delivery (vaginal: cesarean) | 23:5 | 43:8 | 1.00 |
| Nutrition (breast: mixed: formula) | 8:20:0 | 7:44:0 | 0.14 |
| Siblings | 15 (53.6) | 19 (37.3) | 0.24 |
| Use of antibiotics within 1 month before sample collection (%) | 0 (0) | 0 (0) | 1.00 |
| Administration of probiotics or yogurt within 1 month before sample collection (%) (at least two days a week) | 3 (10.7) | 7 (13.7) | 1.00 |
| Allergy-related diseases (%) † | 2 (7.1) | 1 (2.0) | 0.29 |
| fUTI Group (n = 28) | HC Group (n = 51) | |
|---|---|---|
| Phylum | Actinobacceriota | Bacteroidota |
| Class | Actinobacteria | Bacteroidia |
| Bacilli | Negativicutes | |
| Order | Bifidobacteriales | Bacteroidales |
| Enterobacterales | Veillonellases Selenomonadales | |
| Family | Bifidobacteriaceae | Bacteroidaceae |
| Enterobacteriaceae | Veillonellaceae | |
| Genus | Escherichia Shigella | Veillonella |
| Bacteroides |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Urakami, C.; Yamanouchi, S.; Kimata, T.; Tsuji, S.; Akagawa, S.; Kino, J.; Akagawa, Y.; Kato, S.; Araki, A.; Kaneko, K. Abnormal Development of Microbiota May Be a Risk Factor for Febrile Urinary Tract Infection in Infancy. Microorganisms 2023, 11, 2574. https://doi.org/10.3390/microorganisms11102574
Urakami C, Yamanouchi S, Kimata T, Tsuji S, Akagawa S, Kino J, Akagawa Y, Kato S, Araki A, Kaneko K. Abnormal Development of Microbiota May Be a Risk Factor for Febrile Urinary Tract Infection in Infancy. Microorganisms. 2023; 11(10):2574. https://doi.org/10.3390/microorganisms11102574
Chicago/Turabian StyleUrakami, Chika, Sohsaku Yamanouchi, Takahisa Kimata, Shoji Tsuji, Shohei Akagawa, Jiro Kino, Yuko Akagawa, Shogo Kato, Atsushi Araki, and Kazunari Kaneko. 2023. "Abnormal Development of Microbiota May Be a Risk Factor for Febrile Urinary Tract Infection in Infancy" Microorganisms 11, no. 10: 2574. https://doi.org/10.3390/microorganisms11102574