
A Survey of Archaeal Restriction–Modification Systems

Abstract
:1. Introduction
2. Materials and Methods
2.1. Identification of Genomes
2.2. Identification and Clustering of Genes
2.3. Construction of HMM Library
2.4. Bacterial Genes and Genomes
2.5. Characterization of MTase Activity
3. Results and Discussion
3.1. Archaeal Genomes and RM Genes in REBASE
3.2. Functional Categorization of Gene Clusters
3.3. DNA Methylation Phenotypes
3.4. Comparison with Bacteria
3.5. Persistent MTases and RM Systems
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Loenen, W.A.; Dryden, D.T.; Raleigh, E.A.; Wilson, G.G.; Murray, N.E. Highlights of the DNA cutters: A short history of the restriction enzymes. Nucleic Acids Res. 2014, 42, 3–19. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, P.H.; Touchon, M.; Rocha, E.P. The interplay of restriction-modification systems with mobile genetic elements and their prokaryotic hosts. Nucleic Acids Res. 2014, 42, 10618–10631. [Google Scholar] [CrossRef] [PubMed]
- Roberts, R.J.; Vincze, T.; Posfai, J.; Macelis, D. REBASE: A database for DNA restriction and modification: Enzymes, genes and genomes. Nucleic Acids Res. 2023, 51, D629–D630. [Google Scholar] [CrossRef] [PubMed]
- Sayers, E.W.; Cavanaugh, M.; Clark, K.; Pruitt, K.D.; Schoch, C.L.; Sherry, S.T.; Karsch-Mizrachi, I. GenBank. Nucleic Acids Res. 2022, 50, D161–D164. [Google Scholar] [CrossRef]
- McIntyre, A.B.R.; Alexander, N.; Grigorev, K.; Bezdan, D.; Sichtig, H.; Chiu, C.Y.; Mason, C.E. Single-molecule sequencing detection of N6-methyladenine in microbial reference materials. Nat. Commun. 2019, 10, 579. [Google Scholar] [CrossRef]
- Rand, A.C.; Jain, M.; Eizenga, J.M.; Musselman-Brown, A.; Olsen, H.E.; Akeson, M.; Paten, B. Mapping DNA methylation with high-throughput nanopore sequencing. Nat. Methods 2017, 14, 411–413. [Google Scholar] [CrossRef]
- Atack, J.M.; Guo, C.; Litfin, T.; Yang, L.; Blackall, P.J.; Zhou, Y.; Jennings, M.P. Systematic Analysis of REBASE Identifies Numerous Type I Restriction-Modification Systems with Duplicated, Distinct hsdS Specificity Genes That Can Switch System Specificity by Recombination. mSystems 2020, 5, e00497-20. [Google Scholar] [CrossRef]
- Atack, J.M.; Guo, C.; Yang, L.; Zhou, Y.; Jennings, M.P. DNA sequence repeats identify numerous Type I restriction-modification systems that are potential epigenetic regulators controlling phase-variable regulons; phasevarions. FASEB J. 2020, 34, 1038–1051. [Google Scholar] [CrossRef]
- Atack, J.M.; Yang, Y.; Seib, K.L.; Zhou, Y.; Jennings, M.P. A survey of Type III restriction-modification systems reveals numerous, novel epigenetic regulators controlling phase-variable regulons; phasevarions. Nucleic Acids Res. 2018, 46, 3532–3542. [Google Scholar] [CrossRef]
- Ershova, A.S.; Karyagina, A.S.; Vasiliev, M.O.; Lyashchuk, A.M.; Lunin, V.G.; Spirin, S.A.; Alexeevski, A.V. Solitary restriction endonucleases in prokaryotic genomes. Nucleic Acids Res. 2012, 40, 10107–10115. [Google Scholar] [CrossRef]
- Oliveira, P.H.; Fang, G. Conserved DNA Methyltransferases: A Window into Fundamental Mechanisms of Epigenetic Regulation in Bacteria. Trends Microbiol. 2021, 29, 28–40. [Google Scholar] [CrossRef] [PubMed]
- Furuta, Y.; Abe, K.; Kobayashi, I. Genome comparison and context analysis reveals putative mobile forms of restriction-modification systems and related rearrangements. Nucleic Acids Res. 2010, 38, 2428–2443. [Google Scholar] [CrossRef] [PubMed]
- Fullmer, M.S.; Ouellette, M.; Louyakis, A.S.; Papke, R.T.; Gogarten, J.P. The Patchy Distribution of Restriction(-)Modification System Genes and the Conservation of Orphan Methyltransferases in Halobacteria. Genes 2019, 10, 233. [Google Scholar] [CrossRef] [PubMed]
- Blow, M.J.; Clark, T.A.; Daum, C.G.; Deutschbauer, A.M.; Fomenkov, A.; Fries, R.; Froula, J.; Kang, D.D.; Malmstrom, R.R.; Morgan, R.D.; et al. The Epigenomic Landscape of Prokaryotes. PLoS Genet. 2016, 12, e1005854. [Google Scholar] [CrossRef]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef]
- Frickey, T.; Lupas, A. CLANS: A Java application for visualizing protein families based on pairwise similarity. Bioinformatics 2004, 20, 3702–3704. [Google Scholar] [CrossRef]
- Gabler, F.; Nam, S.Z.; Till, S.; Mirdita, M.; Steinegger, M.; Soding, J.; Lupas, A.N.; Alva, V. Protein Sequence Analysis Using the MPI Bioinformatics Toolkit. Curr. Protoc. Bioinform. 2020, 72, e108. [Google Scholar] [CrossRef]
- Malone, T.; Blumenthal, R.M.; Cheng, X. Structure-guided analysis reveals nine sequence motifs conserved among DNA amino-methyltransferases, and suggests a catalytic mechanism for these enzymes. J. Mol. Biol. 1995, 253, 618–632. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE v5 enables improved estimates of phylogenetic tree confidence by ensemble bootstrapping. bioRxiv 2021. [Google Scholar] [CrossRef]
- Yan, B.; Wang, D.; Ettwiller, L. Simultaneous assessment of human genome and methylome data in a single experiment using limited deamination of methylated cytosine. bioRxiv 2023. [Google Scholar] [CrossRef]
- Baum, C.; Lin, Y.C.; Fomenkov, A.; Anton, B.P.; Chen, L.; Yan, B.; Evans, T.C.; Roberts, R.J.; Tolonen, A.C.; Ettwiller, L. Rapid identification of methylase specificity (RIMS-seq) jointly identifies methylated motifs and generates shotgun sequencing of bacterial genomes. Nucleic Acids Res. 2021, 49, e113. [Google Scholar] [CrossRef] [PubMed]
- Marschall, T.; Rahmann, S. Efficient exact motif discovery. Bioinformatics 2009, 25, i356–i364. [Google Scholar] [CrossRef] [PubMed]
- Saad, C.; Noe, L.; Richard, H.; Leclerc, J.; Buisine, M.P.; Touzet, H.; Figeac, M. DiNAMO: Highly sensitive DNA motif discovery in high-throughput sequencing data. BMC Bioinform. 2018, 19, 223. [Google Scholar] [CrossRef] [PubMed]
- Kinch, L.N.; Ginalski, K.; Rychlewski, L.; Grishin, N.V. Identification of novel restriction endonuclease-like fold families among hypothetical proteins. Nucleic Acids Res. 2005, 33, 3598–3605. [Google Scholar] [CrossRef]
- Clark, T.A.; Murray, I.A.; Morgan, R.D.; Kislyuk, A.O.; Spittle, K.E.; Boitano, M.; Fomenkov, A.; Roberts, R.J.; Korlach, J. Characterization of DNA methyltransferase specificities using single-molecule, real-time DNA sequencing. Nucleic Acids Res. 2012, 40, e29. [Google Scholar] [CrossRef]
- Flusberg, B.A.; Webster, D.R.; Lee, J.H.; Travers, K.J.; Olivares, E.C.; Clark, T.A.; Korlach, J.; Turner, S.W. Direct detection of DNA methylation during single-molecule, real-time sequencing. Nat. Methods 2010, 7, 461–465. [Google Scholar] [CrossRef]
- Vaisvila, R.; Ponnaluri, V.K.C.; Sun, Z.; Langhorst, B.W.; Saleh, L.; Guan, S.; Dai, N.; Campbell, M.A.; Sexton, B.S.; Marks, K.; et al. Enzymatic methyl sequencing detects DNA methylation at single-base resolution from picograms of DNA. Genome Res. 2021, 31, 1280–1289. [Google Scholar] [CrossRef]
- Liu, Y.; Siejka-Zielinska, P.; Velikova, G.; Bi, Y.; Yuan, F.; Tomkova, M.; Bai, C.; Chen, L.; Schuster-Bockler, B.; Song, C.X. Bisulfite-free direct detection of 5-methylcytosine and 5-hydroxymethylcytosine at base resolution. Nat. Biotechnol. 2019, 37, 424–429. [Google Scholar] [CrossRef]
- Seshasayee, A.S.; Singh, P.; Krishna, S. Context-dependent conservation of DNA methyltransferases in bacteria. Nucleic Acids Res. 2012, 40, 7066–7073. [Google Scholar] [CrossRef]
- Grogan, D.W. Cytosine methylation by the SuaI restriction-modification system: Implications for genetic fidelity in a hyperthermophilic archaeon. J. Bacteriol. 2003, 185, 4657–4661. [Google Scholar] [CrossRef]
- Couturier, M.; Lindas, A.C. The DNA Methylome of the Hyperthermoacidophilic Crenarchaeon Sulfolobus acidocaldarius. Front. Microbiol. 2018, 9, 137. [Google Scholar] [CrossRef] [PubMed]



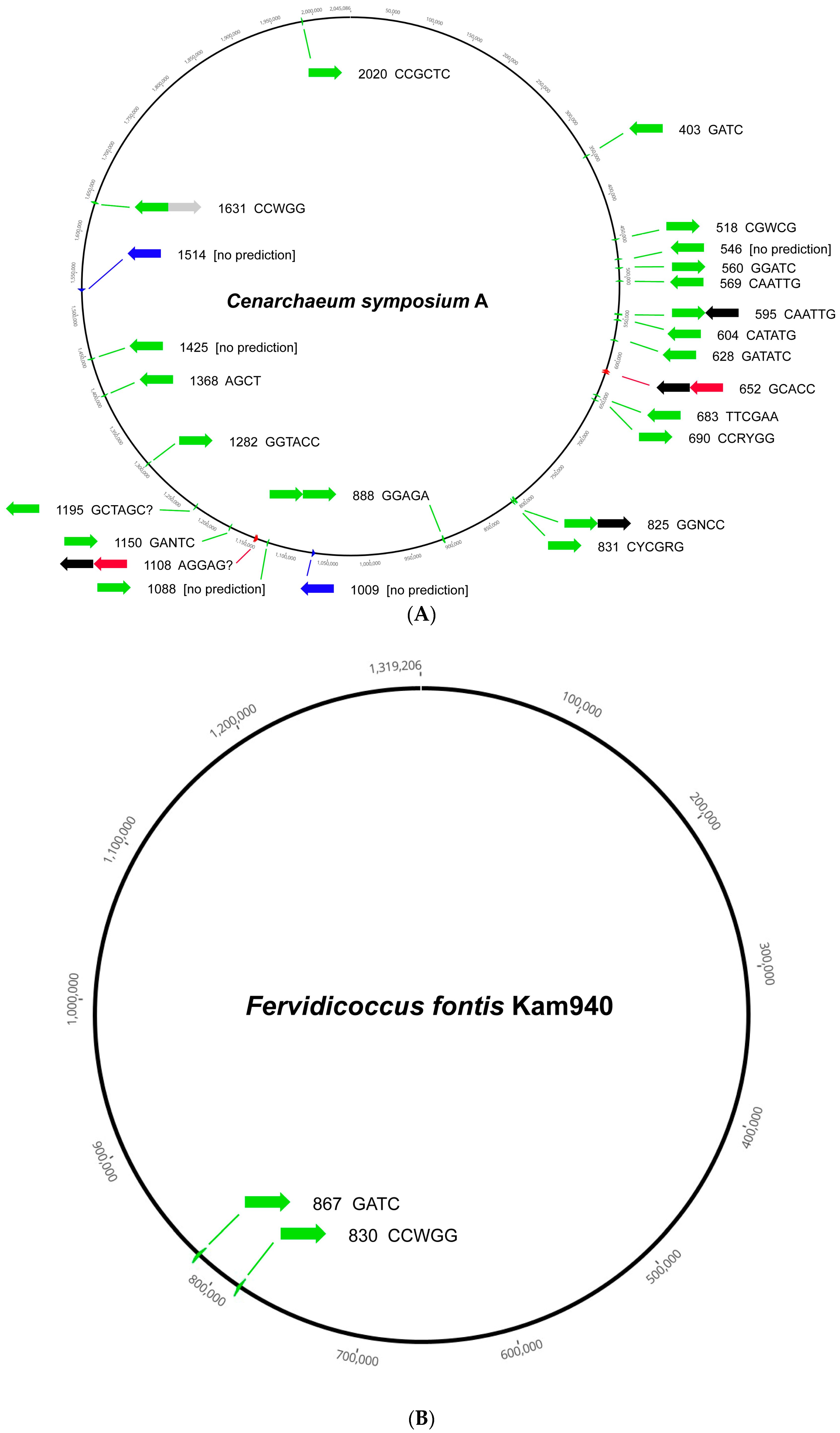{kind=link}
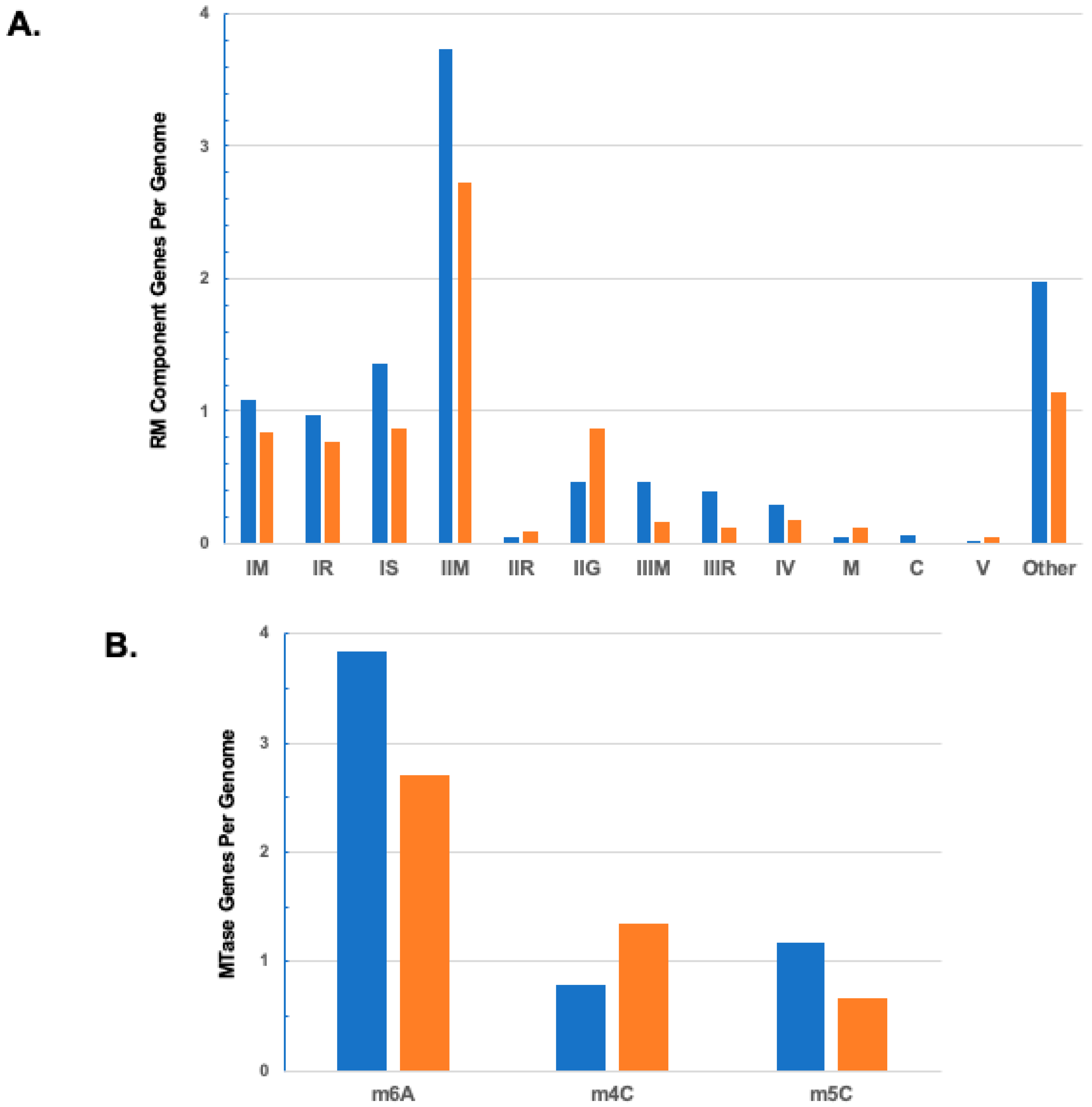{kind=link}
| Taxonomic Group a | No. Genomes | IM | IR | IS | IIM | IIR | IIG | IIIM | IIIR | IV | M | C | V | Other |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Asgard group (Lokiarchaeota) | 1 | 0 | 0 | 0 | 11 | 0 | 4 | 2 | 0 | 0 | 0 | 0 | 0 | 2 |
| Thermoplasmatota | 18 | 0.611 | 0.556 | 0.444 | 3.389 | 0.056 | 0.389 | 0.778 | 0.667 | 0.556 | 0 | 0 | 0.056 | 1.667 |
| Aciduliprofundum | 2 | 0.5 | 0.5 | 0.5 | 1.5 | 0 | 1 | 0.5 | 0.5 | 0 | 0 | 0 | 0 | 1 |
| Thermoplasmata | 16 | 0.625 | 0.562 | 0.438 | 3.625 | 0.062 | 0.312 | 0.812 | 0.688 | 0.625 | 0 | 0 | 0.062 | 1.75 |
| Methanomethylophilaceae | 1 | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 2 |
| Methanomassiliicoccales | 5 | 0.6 | 0.6 | 0.2 | 2 | 0.2 | 0.2 | 0.2 | 0.2 | 1 | 0 | 0 | 0 | 1.4 |
| Thermoplasmatales | 9 | 0.667 | 0.556 | 0.667 | 5 | 0 | 0.444 | 1.333 | 1.111 | 0.333 | 0 | 0 | 0.111 | 1.444 |
| Unclassified | 1 | 0 | 0 | 0 | 3 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 6 |
| Crenarchaeota (Thermoprotei) | 100 | 0.04 | 0.04 | 0.04 | 1.97 | 0.03 | 0.44 | 0.04 | 0.03 | 0 | 0 | 0 | 0.05 | 1.18 |
| Acidilobales | 3 | 0 | 0 | 0 | 1.333 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0.667 |
| Cenarchaeales | 1 | 0 | 0 | 0 | 22 | 0 | 2 | 2 | 2 | 0 | 0 | 0 | 0 | 3 |
| Desulfurococcales | 17 | 0.059 | 0.059 | 0.059 | 2.294 | 0 | 0.647 | 0 | 0 | 0 | 0 | 0 | 0.118 | 0.824 |
| Fervidicoccales | 1 | 0 | 0 | 0 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Sulfolobales | 59 | 0.051 | 0.051 | 0.051 | 1.542 | 0 | 0.39 | 0.034 | 0.017 | 0 | 0 | 0 | 0 | 1.271 |
| Thermofilales | 5 | 0 | 0 | 0 | 2.4 | 0.4 | 0.4 | 0 | 0 | 0 | 0 | 0 | 0.2 | 0.8 |
| Thermoproteales | 14 | 0 | 0 | 0 | 1.929 | 0.071 | 0.429 | 0 | 0 | 0 | 0 | 0 | 0.143 | 1.429 |
| DPANN group | 6 | 0.333 | 0.167 | 0.5 | 1 | 0.167 | 0.167 | 0.5 | 0.333 | 0 | 0 | 0 | 0 | 0.167 |
| Micrarchaeota | 2 | 0 | 0 | 0.5 | 1.5 | 0.5 | 0 | 1.5 | 1 | 0 | 0 | 0 | 0 | 0 |
| Nanohaloarchaeota (Nanohalobia) | 1 | 1 | 1 | 1 | 1 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 1 |
| Nanoarchaeota | 3 | 0.333 | 0 | 0.333 | 0.667 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Environmental sample | 1 | 1 | 1 | 1 | 9 | 0 | 1 | 1 | 1 | 0 | 0 | 0 | 0 | 0 |
| Euryarchaeota | 362 | 1.124 | 1.033 | 1.157 | 2.798 | 0.113 | 1.064 | 0.152 | 0.130 | 0.227 | 0.180 | 0 | 0.055 | 1.174 |
| Archaeoglobi (Archaeoglobales) | 8 | 1 | 0.75 | 0.875 | 0.875 | 0 | 0.25 | 0.25 | 0.25 | 0 | 0 | 0 | 0 | 1.375 |
| Methanoliparia | 1 | 2 | 2 | 2 | 5 | 0 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 3 |
| Methanomada | 61 | 1 | 0.951 | 1.344 | 2.082 | 0.262 | 0.77 | 0.41 | 0.328 | 0.279 | 0.148 | 0 | 0.033 | 1.656 |
| Methanobacteria | 36 | 0.944 | 0.889 | 1.472 | 1.444 | 0.333 | 0.611 | 0.361 | 0.278 | 0.472 | 0.222 | 0 | 0.056 | 1.222 |
| Methanococci | 24 | 1.125 | 1.083 | 1.208 | 3.083 | 0.167 | 0.958 | 0.5 | 0.417 | 0 | 0.042 | 0 | 0 | 2.375 |
| Methanopyri | 1 | 0 | 0 | 0 | 1 | 0 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Methanonatronarchaeia | 1 | 0 | 0 | 0 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 |
| Halobacteria | 181 | 0.431 | 0.409 | 0.420 | 3.602 | 0.11 | 1.354 | 0.033 | 0.028 | 0.066 | 0.149 | 0 | 0.055 | 0.890 |
| Halobacteriales | 94 | 0.457 | 0.426 | 0.415 | 3.320 | 0.128 | 1.362 | 0.011 | 0.011 | 0.053 | 0.138 | 0 | 0.064 | 0.904 |
| Haloferacales | 45 | 0.444 | 0.422 | 0.489 | 3.533 | 0.089 | 1.4 | 0.067 | 0.044 | 0.133 | 0.089 | 0 | 0.067 | 0.844 |
| Natrialbales | 42 | 0.357 | 0.357 | 0.357 | 4.31 | 0.095 | 1.286 | 0.048 | 0.048 | 0.024 | 0.238 | 0 | 0.024 | 0.905 |
| Methanomicrobia | 65 | 3.585 | 3.215 | 3.462 | 2.462 | 0.015 | 0.831 | 0.323 | 0.292 | 0.815 | 0.446 | 0 | 0.077 | 1.138 |
| Methanocellales | 3 | 1 | 1 | 1 | 6 | 0 | 0.333 | 0.667 | 0.333 | 0 | 0 | 0 | 0 | 1.333 |
| Methanomicrobiales | 19 | 2.105 | 1.684 | 1.789 | 4.579 | 0.053 | 1.105 | 0.368 | 0.316 | 0.737 | 0.421 | 0 | 0.053 | 1.789 |
| Methanosarcinales | 42 | 4.524 | 4.143 | 4.476 | 1.143 | 0 | 0.738 | 0.286 | 0.286 | 0.905 | 0.5 | 0 | 0.095 | 0.833 |
| Unclassified | 1 | 0 | 0 | 0 | 7 | 0 | 1 | 0 | 0 | 1 | 0 | 0 | 0 | 1 |
| Nanohaloarchaeota | 1 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Thermococci (Thermococcales) | 44 | 0.568 | 0.568 | 0.614 | 1.341 | 0.091 | 0.795 | 0.023 | 0.023 | 0 | 0 | 0 | 0.045 | 1.705 |
| TACK group | 31 | 0.419 | 0.419 | 0.581 | 3.71 | 0 | 0.29 | 0.129 | 0 | 0 | 0 | 0 | 0.097 | 0.516 |
| Geothermarchaeota | 1 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 1 |
| Korarchaeota | 1 | 0 | 0 | 0 | 1 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Nitrososphaerota | 29 | 0.448 | 0.448 | 0.621 | 3.931 | 0 | 0.241 | 0.103 | 0 | 0 | 0 | 0 | 0.103 | 0.517 |
| Nitrosopumilales | 14 | 0.286 | 0.286 | 0.571 | 2.786 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0.714 |
| Nitrososphaeria | 10 | 0.6 | 0.6 | 0.6 | 5.8 | 0 | 0.7 | 0.2 | 0 | 0 | 0 | 0 | 0.3 | 0.1 |
| Nitrososphaerota inc. sed. | 5 | 0.6 | 0.6 | 0.8 | 3.4 | 0 | 0 | 0.2 | 0 | 0 | 0 | 0 | 0 | 0.8 |
| TOTAL | 519 | 0.844 | 0.776 | 0.873 | 2.721 | 0.089 | 0.869 | 0.160 | 0.125 | 0.177 | 0.125 | 0 | 0.056 | 1.141 |
| All Complete Genomes | Genomes with Methylation Data | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Taxonomic Group a | Genomes | Genes m6A | Genes m4C | Genes m5C | Genomes | Genes m6A | Genes m4C | Genes m5C | Motifs m6A | Motifs m4C | Motifs m5C |
| Asgard group (Lokiarchaeota) | 1 | 10 | 7 | 0 | |||||||
| Thermoplasmatota | 18 | 2.556 | 2.167 | 0.444 | 2 | 6 | 4.5 | 0 | 5 | 2.5 | 0 |
| Aciduliprofundum | 2 | 2 | 1.5 | 0 | |||||||
| Thermoplasmata | 16 | 2.625 | 2.25 | 0.5 | |||||||
| Methanomethylophilaceae | 1 | 1 | 0 | 0 | |||||||
| Methanomassiliicoccales | 5 | 1.4 | 1 | 0.6 | |||||||
| Thermoplasmatales | 9 | 3.778 | 3.444 | 0.222 | 2 | 6 | 4.5 | 0 | 5 | 2.5 | 0 |
| Unclassified | 1 | 0 | 0 | 3 | |||||||
| Crenarchaeota (Thermoprotei) | 100 | 0.83 | 0.56 | 1.1 | 3 | 0.667 | 0.667 | 1 | 1 | 0.667 | 0.667 |
| Acidilobales | 3 | 0.333 | 0 | 1 | 1 | 0 | 0 | 1 | 0 | 0 | 1 |
| Cenarchaeales | 1 | 13 | 12 | 1 | |||||||
| Desulfurococcales | 17 | 1 | 0.882 | 1.118 | 1 | 2 | 1 | 1 | 3 | 1 | 0 |
| Fervidicoccales | 1 | 0 | 1 | 1 | |||||||
| Sulfolobales | 59 | 0.576 | 0.441 | 1 | 1 | 0 | 1 | 1 | 0 | 1 | 1 |
| Thermofilales | 5 | 1 | 0.4 | 1.4 | |||||||
| Thermoproteales | 14 | 0.929 | 0 | 1.429 | |||||||
| DPANN group | 6 | 1.333 | 0.5 | 0.167 | 1 | 1 | 0 | 0 | 1 | 0 | 0 |
| Micrarchaeota | 2 | 2 | 1 | 0 | |||||||
| Nanohaloarchaeota (Nanohalobia) | 1 | 2 | 0 | 1 | |||||||
| Nanoarchaeota | 3 | 0.667 | 0.333 | 0 | 1 | 1 | 0 | 0 | 1 | 0 | 0 |
| Environmental sample | 1 | 6 | 6 | 0 | |||||||
| Euryarchaeota | 362 | 3.282 | 1.442 | 0.594 | 39 | 3.538 | 1.615 | 0.641 | 2.538 | 1.308 | 0.103 |
| Archaeoglobi (Archaeoglobales) | 8 | 1.625 | 0.375 | 0.375 | |||||||
| Methanoliparia | 1 | 4 | 5 | 0 | |||||||
| Methanomada | 61 | 3.033 | 0.574 | 0.803 | 5 | 4 | 1.2 | 1.2 | 3.4 | 1 | 0.2 |
| Methanobacteria | 36 | 2.806 | 0.25 | 0.528 | 3 | 3 | 0.333 | 1 | 2.333 | 0.333 | 0 |
| Methanococci | 24 | 3.417 | 1.083 | 1.208 | 2 | 5.5 | 2.5 | 1.5 | 5 | 2 | 0.5 |
| Methanopyri | 1 | 2 | 0 | 1 | |||||||
| Methanonatronarchaeia | 1 | 0 | 1 | 1 | |||||||
| Halobacteria | 181 | 2.856 | 2.055 | 0.657 | 26 | 3.462 | 1.769 | 0.615 | 2.461 | 1.269 | 0.077 |
| Halobacteriales | 94 | 2.777 | 1.872 | 0.638 | 10 | 3.1 | 1.2 | 0.4 | 1.6 | 1.1 | 0.1 |
| Haloferacales | 45 | 3.2 | 1.556 | 0.778 | 10 | 3.5 | 1.5 | 0.9 | 2.9 | 1.1 | 0.1 |
| Natrialbales | 42 | 2.667 | 3 | 0.571 | 6 | 4 | 3.167 | 0.5 | 3.167 | 1.833 | 0 |
| Methanomicrobia | 65 | 6.031 | 1.277 | 0.338 | 4 | 5 | 2.5 | 0.25 | 2.5 | 3 | 0.25 |
| Methanocellales | 3 | 4.667 | 2.667 | 0.667 | |||||||
| Methanomicrobiales | 19 | 5.316 | 2.895 | 0.368 | 3 | 6 | 3 | 0.333 | 3 | 3.333 | 0.333 |
| Methanosarcinales | 42 | 6.548 | 0.381 | 0.262 | 1 | 2 | 1 | 0 | 1 | 2 | 0 |
| Unclassified | 1 | 2 | 4 | 2 | |||||||
| Nanohaloarchaeota | 1 | 0 | 1 | 0 | |||||||
| Thermococci (Thermococcales) | 44 | 1.75 | 0.5 | 0.477 | 4 | 2 | 0.25 | 0.5 | 2 | 0.25 | 0 |
| TACK group | 31 | 2.065 | 2.129 | 0.355 | 4 | 1.5 | 1.5 | 0.5 | 1.5 | 1.25 | 0.25 |
| Geothermarchaeota | 1 | 2 | 0 | 0 | |||||||
| Korarchaeota | 1 | 1 | 1 | 0 | |||||||
| Nitrososphaerota | 29 | 2.103 | 2.241 | 0.379 | 4 | 1.5 | 1.5 | 0.5 | 1.5 | 1.25 | 0.25 |
| Nitrosopumilales | 14 | 1.643 | 1.071 | 0.357 | 3 | 1.667 | 1 | 0.667 | 1.667 | 1 | 0.333 |
| Nitrososphaeria | 10 | 2.8 | 4.1 | 0.4 | 1 | 1 | 3 | 0 | 1 | 2 | 0 |
| Nitrososphaerota inc. sed. | 5 | 2 | 1.8 | 0.4 | |||||||
| TOTAL | 519 | 2.707 | 1.347 | 0.665 | 49 | 3.245 | 1.633 | 0.612 | 2.429 | 1.286 | 0.143 |
| Taxonomic Group a | Total Genomes | Cluster (Members) | Class | Motif b |
|---|---|---|---|---|
| Thermoplasmatota | 18 | None | ||
| Crenarchaeota (Thermoprotei) | 100 | HG2 (99) | IIM | CCWGG (m5C) |
| Sulfolobales | 59 | None | ||
| Sulfolobus acidocaldarius | 9 | HG15 M/R (8) | IIM/R | GGCC (m4C) |
| DPANN group | 6 | None | ||
| Euryarchaeota | 363 | None | ||
| Archaeoglobi (Archaeoglobales) | 8 | HG6 M/R (6) | IM/R | n/d |
| Methanomada | 61 | None | ||
| Methanobacteria | 36 | None | ||
| Methanobacterium | 12 | HG3 (10) | IIM | GATC (m6A) |
| Methanococci | 24 | HG2 (19) | IIM | CCWGG (m5C) |
| Methanococcus maripaludis | 9 | HG3 (9) | IIM | GATC (m6A) |
| Halobacteria | 182 | HG1 (155) | IIM | CTAG (m4C) |
| Haloferacales | 45 | None | ||
| Halorubraceae | 20 | HG3 (15) | IIM | GATC (m6A) |
| Natrialbales | 42 | HG4 (41) | IIM | CATTC (m6A) |
| Haloterrigena | 9 | HG5 (7) | BREX | CTGGAG (m6A) |
| Methanomicrobia | 65 | None | ||
| Methanomicrobiales | 19 | HG3 (15) | IIM | GATC (m6A) |
| Methanoculleus | 6 | HG16 (6) | IIM | AGCT (m4C) |
| M-regula/M-spirilla group c | 6 | HG16 (6) | IIM | AGCT (m4C) |
| M-regula/M-spirilla group c | 6 | HG18 (6) | IIM | GTAC (m4C) |
| M-regula/M-spirilla group c | 6 | HG20 (6) | IIM | CTNAG (m4C) |
| Methanosarcinales | 42 | HG8 M/R/S (28) | IM/R/S | n/d |
| Methanosarcina | 29 | HG9 (22) | IV | n/d |
| Methanosarcina mazei | 9 | HG17 M/R (7) | IM/R | n/d |
| Methanosarcina mazei | 9 | HG13 (7) | BREX | n/d |
| Methanosarcina mazei | 9 | HG14 M/R (9) | IM/R | n/d |
| Methanosarcina mazei | 9 | HG7 M/R/S (9) | IM/R/S | n/d |
| Thermococci (Thermococcales) | 44 | None | ||
| Pyrococcus | 9 | HG2 (9) | IIM | CCWGG (m5C) |
| TACK group | 31 | None | ||
| Nitrososphaerota | 29 | HG3 (27) | IIM | GATC (m6A) |
| Nitrosopumilales | 14 | HG11 (14) | IIM | AGCT (m4C) |
| Nitrososphaeria | 10 | |||
| Nitrososphaerales | 8 | HG10 (8) | IIM | GTAC (m4C) |
| Nitrososphaerales | 8 | HG19 (8) | IIM | AGCT (m4C) |
| Nitrososphaera | 5 | HG12 (5) | IIM | CGCG (m4C) |
| Nitrososphaera | 5 | HG21 (5) | IIM | Unknown |
| Nitrososphaerota inc. sed. | 5 | HG11 (4) | IIM | AGCT (m4C) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Anton, B.P.; Roberts, R.J. A Survey of Archaeal Restriction–Modification Systems. Microorganisms 2023, 11, 2424. https://doi.org/10.3390/microorganisms11102424
Anton BP, Roberts RJ. A Survey of Archaeal Restriction–Modification Systems. Microorganisms. 2023; 11(10):2424. https://doi.org/10.3390/microorganisms11102424
Chicago/Turabian StyleAnton, Brian P., and Richard J. Roberts. 2023. "A Survey of Archaeal Restriction–Modification Systems" Microorganisms 11, no. 10: 2424. https://doi.org/10.3390/microorganisms11102424