
Atlantic Salmon (Salmo salar) Transfer to Seawater by Gradual Salinity Changes Exhibited an Increase in The Intestinal Microbial Abundance and Richness

 ,
, 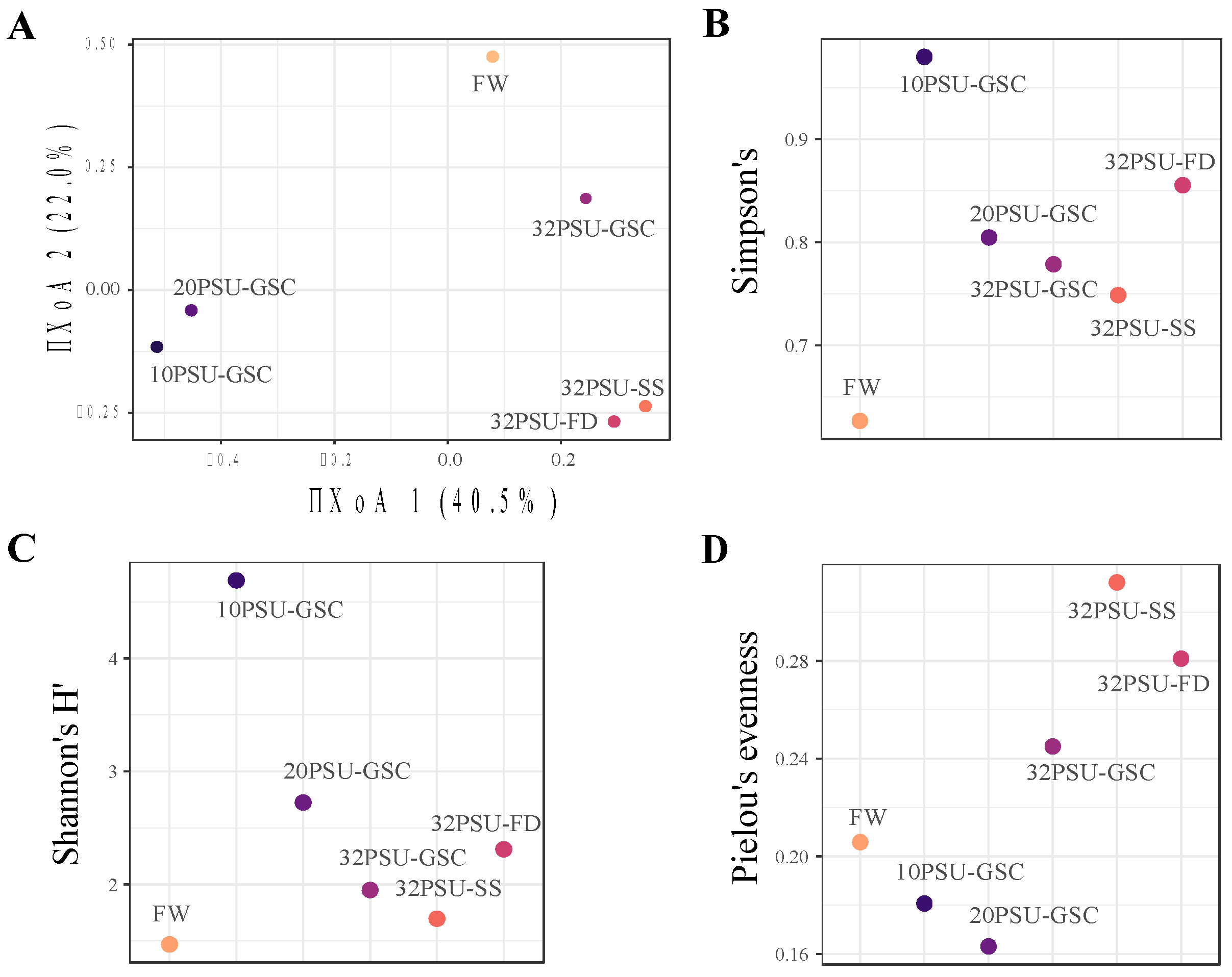{kind=link}
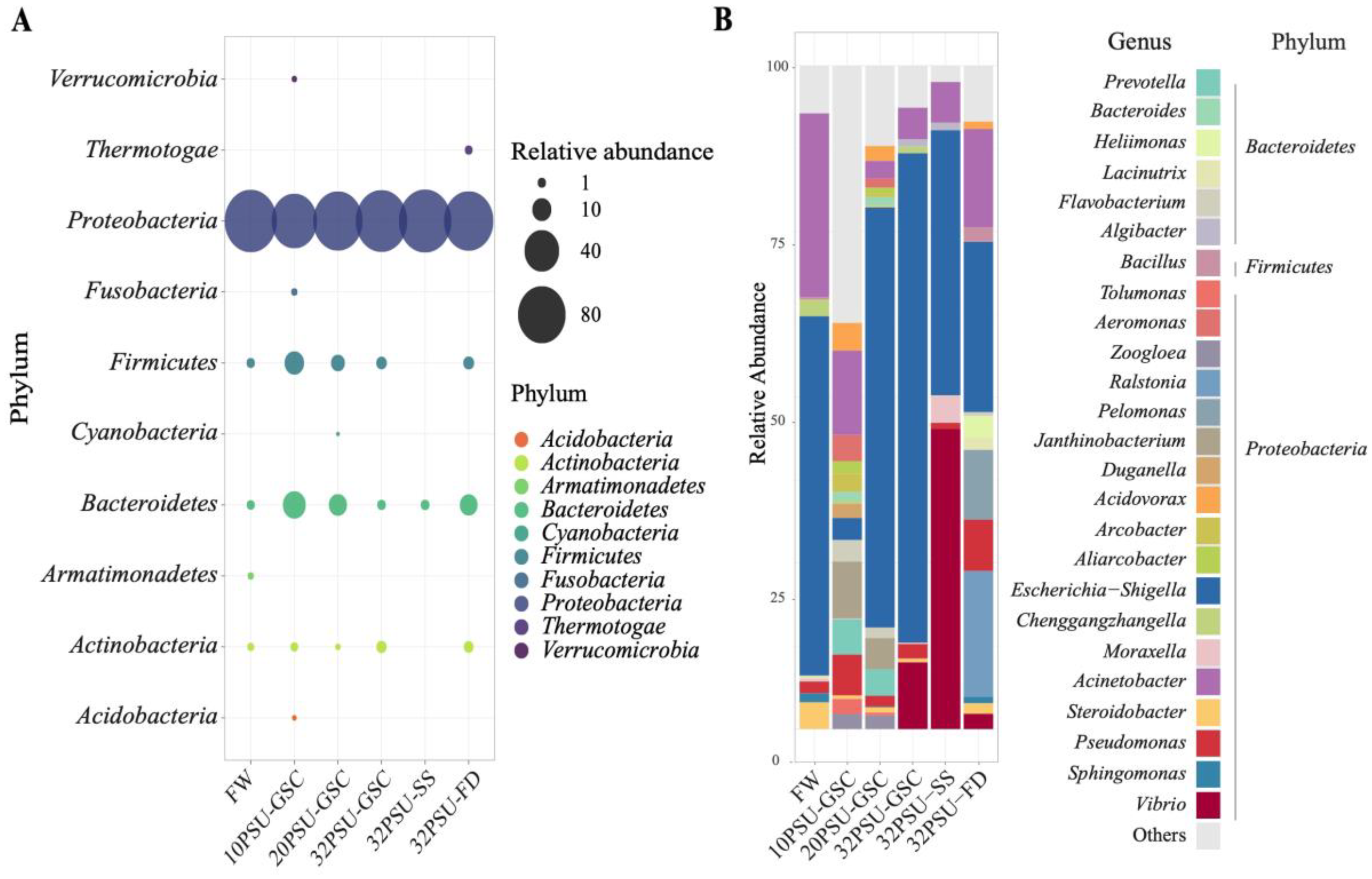{kind=link}
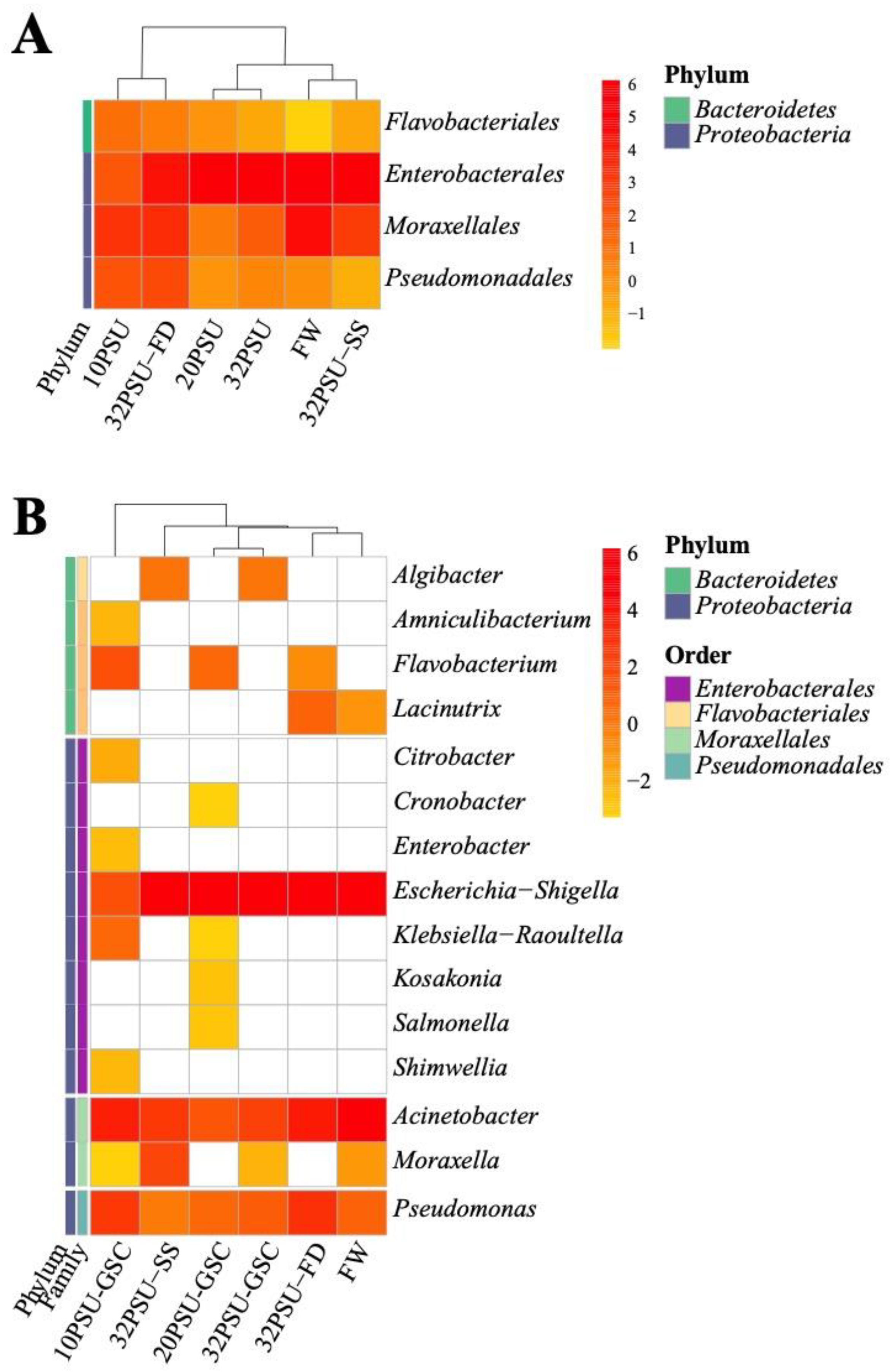{kind=link}
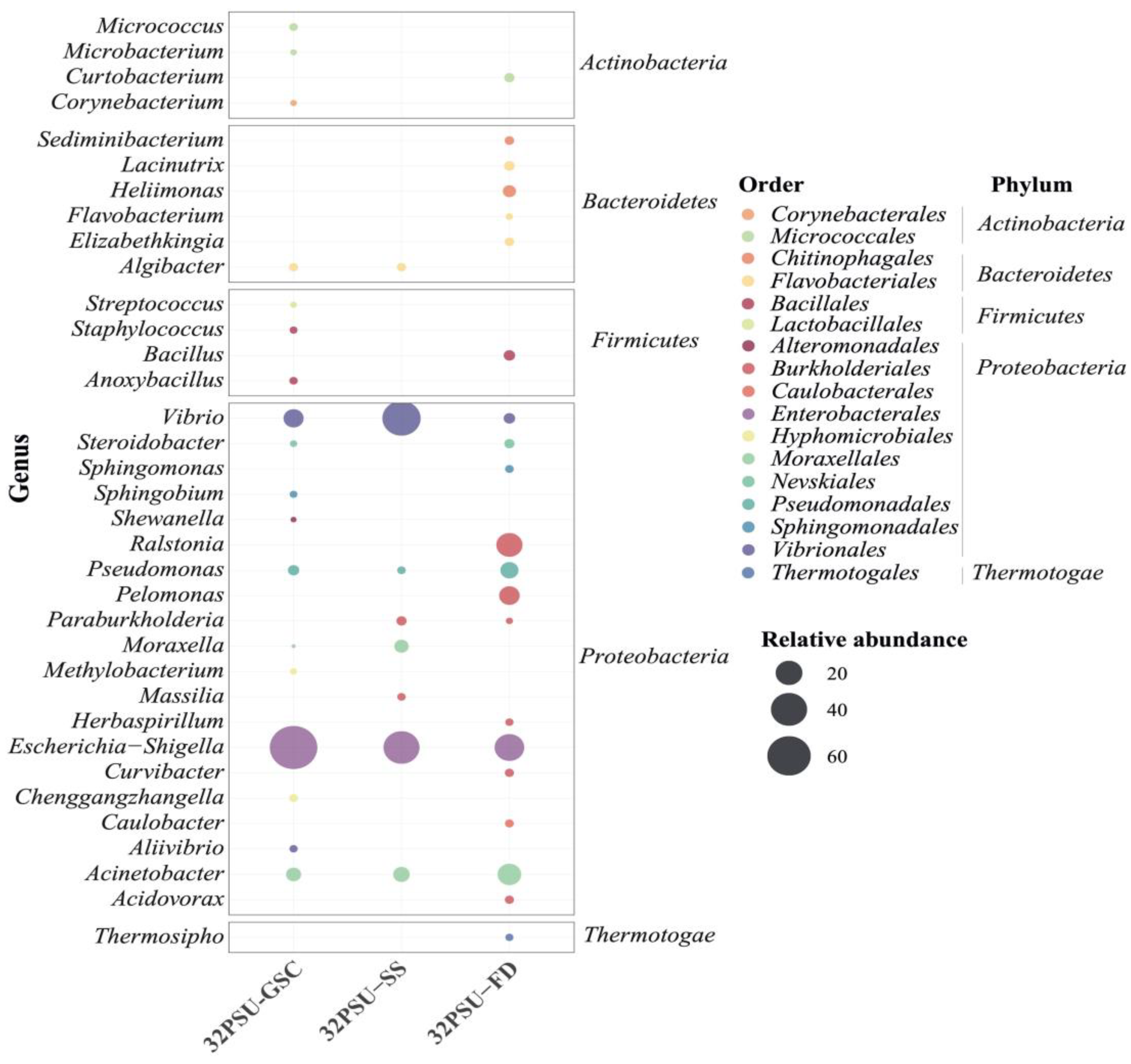{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Experimental Design
2.2. DNA Extraction and Full-16S rRNA Amplification
2.3. Nanopore Library Synthesis and Sequencing
2.4. Bioinformatic Analysis
3. Results
3.1. Experimental Performance of Atlantic Salmon
3.2. Full-16S Sequencing Data Report
3.3. Structure of The Intestinal Microbial Community
3.4. Atlantic Salmon Intestine Microbiota Composition during The SW Transfer
3.5. Bacteria Species Identified in the Intestinal Atlantic Salmon Microbiota
3.6. Intestine Microbiota after Seawater Transfer
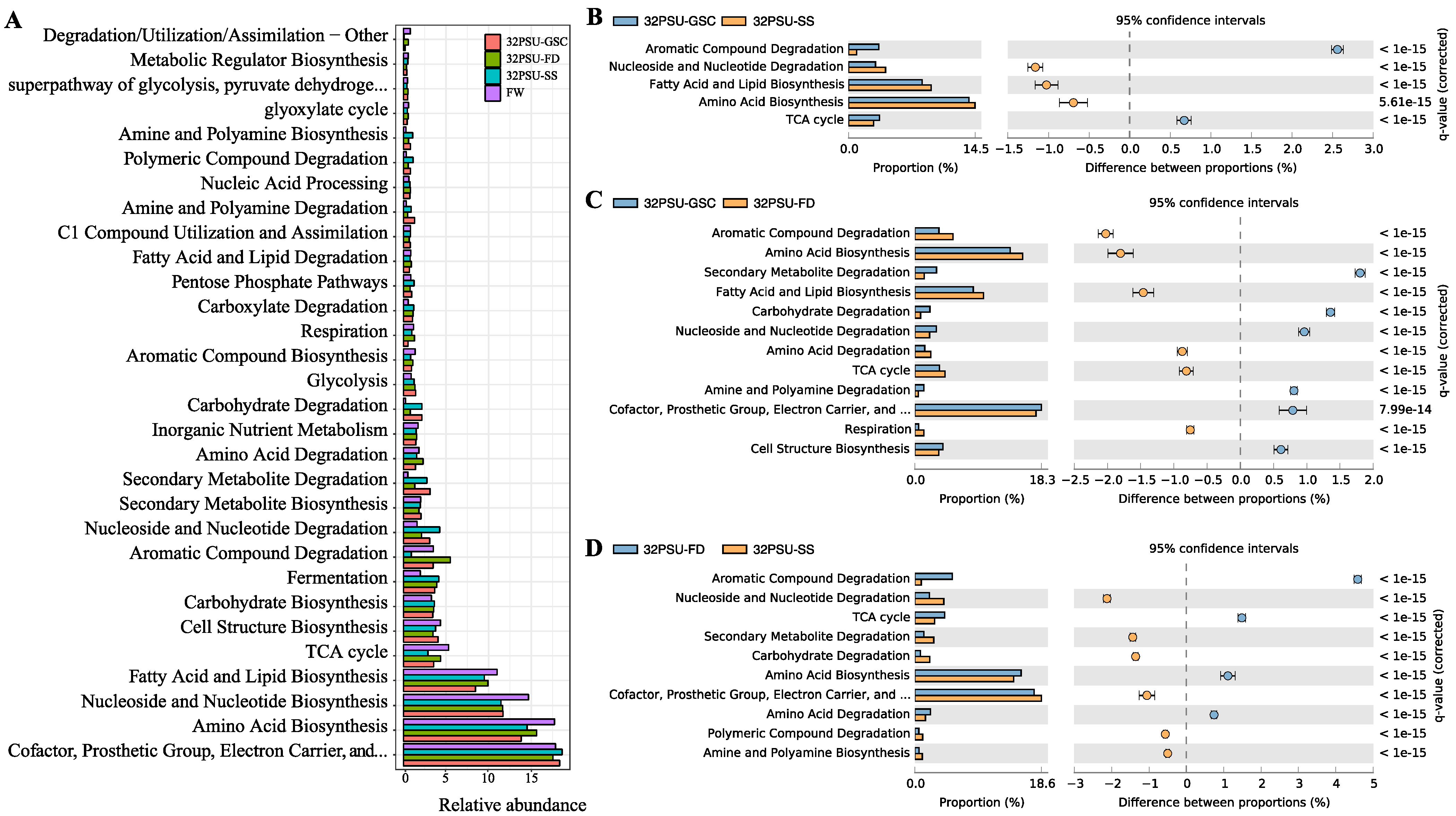
3.7. Functional Analysis in the Intestinal Atlantic Salmon Microbiota
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Egerton, S.; Culloty, S.; Whooley, J.; Stanton, C.; Ross, R.P. The Gut Microbiota of Marine Fish. Front. Microbiol. 2018, 9, 873. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, V.T.; Smith, K.F.; Melvin, D.W.; Amaral-Zettler, L.A. Community assembly of a euryhaline fish microbiome during salinity acclimation. Mol. Ecol. 2015, 24, 2537–2550. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Bruni, L.; Jaramillo-Torres, A.; Gajardo, K.; Kortner, T.M.; Krogdahl, A. Differential response of digesta- and mucosa-associated intestinal microbiota to dietary insect meal during the seawater phase of Atlantic salmon. Anim. Microbiome 2021, 3, 8. [Google Scholar] [CrossRef]
- Harrison, B.K.; Myrbo, A.; Flood, B.E.; Bailey, J.V. Abrupt burial imparts persistent changes to the bacterial diversity of turbidite-associated sediment profiles. Geobiology 2018, 16, 190–202. [Google Scholar] [CrossRef] [PubMed]
- Labonte, J.M.; Lever, M.A.; Edwards, K.J.; Orcutt, B.N. Influence of Igneous Basement on Deep Sediment Microbial Diversity on the Eastern Juan de Fuca Ridge Flank. Front. Microbiol. 2017, 8, 1434. [Google Scholar] [CrossRef] [Green Version]
- Mills, H.J.; Reese, B.K.; Shepard, A.K.; Riedinger, N.; Dowd, S.E.; Morono, Y.; Inagaki, F. Characterization of Metabolically Active Bacterial Populations in Subseafloor Nankai Trough Sediments above, within, and below the Sulfate-Methane Transition Zone. Front. Microbiol. 2012, 3, 113. [Google Scholar] [CrossRef] [Green Version]
- Rud, I.; Kolarevic, J.; Holan, A.B.; Berget, I.; Calabrese, S.; Terjesen, B.F. Deep-sequencing of the bacterial microbiota in commercial-scale recirculating and semi-closed aquaculture systems for Atlantic salmon post-smolt production. Aquac. Eng. 2017, 78, 50–62. [Google Scholar] [CrossRef]
- Lokesh, J.; Kiron, V. Transition from freshwater to seawater reshapes the skin-associated microbiota of Atlantic salmon. Sci. Rep. 2016, 6, 19707. [Google Scholar] [CrossRef] [Green Version]
- Lorgen-Ritchie, M.; Clarkson, M.; Chalmers, L.; Taylor, J.F.; Migaud, H.; Martin, S.A.M. A Temporally Dynamic Gut Microbiome in Atlantic Salmon During Freshwater Recirculating Aquaculture System (RAS) Production and Post-seawater Transfer. Front. Mar. Sci. 2021, 8, 711797. [Google Scholar] [CrossRef]
- Wang, J.; Jaramillo-Torres, A.; Li, Y.; Kortner, T.M.; Gajardo, K.; Brevik, O.J.; Jakobsen, J.V.; Krogdahl, A. Microbiota in intestinal digesta of Atlantic salmon (Salmo salar), observed from late freshwater stage until one year in seawater, and effects of functional ingredients: A case study from a commercial sized research site in the Arctic region. Anim. Microbiome 2021, 3, 14. [Google Scholar] [CrossRef]
- Bukin, Y.S.; Galachyants, Y.P.; Morozov, I.V.; Bukin, S.V.; Zakharenko, A.S.; Zemskaya, T.I. The effect of 16S rRNA region choice on bacterial community metabarcoding results. Sci. Data 2019, 6, 190007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kerrigan, Z.; Kirkpatrick, J.B.; D'Hondt, S. Influence of 16S rRNA Hypervariable Region on Estimates of Bacterial Diversity and Community Composition in Seawater and Marine Sediment. Front. Microbiol. 2019, 10, 1640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Willis, C.; Desai, D.; LaRoche, J. Influence of 16S rRNA variable region on perceived diversity of marine microbial communities of the Northern North Atlantic. Fems Microbiol. Lett. 2019, 366, fnz152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yarza, P.; Yilmaz, P.; Pruesse, E.; Glockner, F.O.; Ludwig, W.; Schleifer, K.H.; Whitman, W.B.; Euzeby, J.; Amann, R.; Rossello-Mora, R. Uniting the classification of cultured and uncultured bacteria and archaea using 16S rRNA gene sequences. Nat. Rev. Microbiol. 2014, 12, 635–645. [Google Scholar] [CrossRef]
- Matsuo, Y.; Komiya, S.; Yasumizu, Y.; Yasuoka, Y.; Mizushima, K.; Takagi, T.; Kryukov, K.; Fukuda, A.; Morimoto, Y.; Naito, Y.; et al. Full-length 16S rRNA gene amplicon analysis of human gut microbiota using MinION nanopore sequencing confers species-level resolution. BMC Microbiol. 2021, 21, 35. [Google Scholar] [CrossRef]
- Fujiyoshi, S.; Muto-Fujita, A.; Maruyama, F. Evaluation of PCR conditions for characterizing bacterial communities with full-length 16S rRNA genes using a portable nanopore sequencer. Sci. Rep. 2020, 10, 12580. [Google Scholar] [CrossRef]
- Johnson, J.S.; Spakowicz, D.J.; Hong, B.Y.; Petersen, L.M.; Demkowicz, P.; Chen, L.; Leopold, S.R.; Hanson, B.M.; Agresta, H.O.; Gerstein, M.; et al. Evaluation of 16S rRNA gene sequencing for species and strain-level microbiome analysis. Nat. Commun. 2019, 10, 5029. [Google Scholar] [CrossRef] [Green Version]
- Shin, J.; Lee, S.; Go, M.J.; Lee, S.Y.; Kim, S.C.; Lee, C.H.; Cho, B.K. Analysis of the mouse gut microbiome using full-length 16S rRNA amplicon sequencing. Sci. Rep. 2016, 6, 29681. [Google Scholar] [CrossRef] [Green Version]
- Cusco, A.; Catozzi, C.; Vines, J.; Sanchez, A.; Francino, O. Microbiota profiling with long amplicons using Nanopore sequencing: Full-length 16S rRNA gene and the 16S-ITS-23S of the rrn operon. F1000Res 2018, 7, 1755. [Google Scholar] [CrossRef]
- Dehler, C.E.; Secombes, C.J.; Martin, S.A. Environmental and physiological factors shape the gut microbiota of Atlantic salmon parr (Salmo salar L.). Aquaculture 2017, 467, 149–157. [Google Scholar] [CrossRef]
- Skrodenyte-Arbaciauskiene, V.; Virbickas, T.; Luksa, J.; Serviene, E.; Blazyte-Cereskiene, L.; Kesminas, V. Gut Microbiome of Wild Baltic Salmon (Salmo salar L.) Parr. Microb. Ecol. 2022, 84, 1294–1298. [Google Scholar] [CrossRef] [PubMed]
- Sprockett, D.; Fukami, T.; Relman, D.A. Role of priority effects in the early-life assembly of the gut microbiota. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 197–205. [Google Scholar] [CrossRef] [PubMed]
- Dulski, T.; Kujawa, R.; Godzieba, M.; Ciesielski, S. Effect of Salinity on the Gut Microbiome of Pike Fry (Esox lucius). Appl. Sci. 2020, 10, 2506. [Google Scholar] [CrossRef] [Green Version]
- Wu, P.; Liu, Y.; Li, C.; Xiao, Y.; Wang, T.; Lin, L.; Xie, Y. The Composition of Intestinal Microbiota From Collichthys lucidus and Its Interaction With Microbiota From Waters Along the Pearl River Estuary in China. Front. Environ. Sci. 2021, 9, 675856. [Google Scholar] [CrossRef]
- Sundh, H.; Nilsen, T.O.; Lindstrom, J.; Hasselberg-Frank, L.; Stefansson, S.O.; McCormick, S.D.; Sundell, K. Development of intestinal ion-transporting mechanisms during smoltification and seawater acclimation in Atlantic salmon Salmo salar. J. Fish Biol. 2014, 85, 1227–1252. [Google Scholar] [CrossRef]
- Dehler, C.E.; Secombes, C.J.; Martin, S.A.M. Seawater transfer alters the intestinal microbiota profiles of Atlantic salmon (Salmo salar L.). Sci. Rep. 2017, 7, 13877. [Google Scholar] [CrossRef] [Green Version]
- Milligan-Myhre, K.; Small, C.M.; Mittge, E.K.; Agarwal, M.; Currey, M.; Cresko, W.A.; Guillemin, K. Innate immune responses to gut microbiota differ between oceanic and freshwater threespine stickleback populations. Dis. Model Mech. 2016, 9, 187–198. [Google Scholar] [CrossRef] [Green Version]
- Sernapesca. Informe Sanitario con Informacion Sanitaria de Agua Dulce y Mar Año 2021; Departamento de Salud Animal, Subdirección de Acuicultura, Gobierno de Chile: Santiago, Chile, 2021. [Google Scholar]
- Nisembaum, L.G.; Martin, P.; Lecomte, F.; Falcon, J. Melatonin and osmoregulation in fish: A focus on Atlantic salmon Salmo salar smoltification. J. Neuroendocrinol. 2021, 33, e12955. [Google Scholar] [CrossRef]
- Folmar, L.C.; Dickhoff, W.W. The parr-smolt transformation (smotification) and seawater adaptation in salmonids. A review of selected literature. Aquaculture 1980, 21, 1–37. [Google Scholar] [CrossRef]
- Johansson, L.H.; Timmerhaus, G.; Afanasyev, S.; Jorgensen, S.M.; Krasnov, A. Smoltification and seawater transfer of Atlantic salmon (Salmo salar L.) is associated with systemic repression of the immune transcriptome. Fish Shellfish Immunol. 2016, 58, 33–41. [Google Scholar] [CrossRef]
- Sahlmann, C.; Djordjevic, B.; Lagos, L.; Mydland, L.T.; Morales-Lange, B.; Øvrum Hansen, J.; Ånestad, R.; Mercado, L.; Bjelanovic, M.; Press, C.M.; et al. Yeast as a protein source during smoltification of Atlantic salmon (Salmo salar L.), enhances performance and modulates health. Aquaculture 2019, 513, 734396. [Google Scholar] [CrossRef]
- Valenzuela-Muñoz, V.; Gallardo-Escarate, C.; Benavente, B.P.; Valenzuela-Miranda, D.; Nunez-Acuna, G.; Escobar-Sepulveda, H.; Valdes, J.A. Whole-Genome Transcript Expression Profiling Reveals Novel Insights into Transposon Genes and Non-Coding RNAs during Atlantic Salmon Seawater Adaptation. Biology 2021, 11, 1. [Google Scholar] [CrossRef] [PubMed]
- Lane, D.J. 16S/23S rRNA sequencing. In Nucleic Acid Techniques in Bacterial Systematics; Stackebrandt, E., Goodfellow, M., Eds.; Wiley: Hoboken, NJ, USA, 1991; Volume 4, pp. 115–175. [Google Scholar]
- Wick, R. Porechop: An adapter trimmer for Oxford Nanopore reads. 2018. Available online: https://github.com/rrwick/Porechop (accessed on 15 July 2022).
- Rodriguez-Perez, H.; Ciuffreda, L.; Flores, C. NanoCLUST: A species-level analysis of 16S rRNA nanopore sequencing data. Bioinformatics 2021, 37, 1600–1601. [Google Scholar] [CrossRef] [PubMed]
- Dixon, P. VEGAN, a package of R functions for community ecology. J. Veg. Sci. 2003, 14, 927–930. [Google Scholar] [CrossRef]
- R-Core-Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2020. [Google Scholar]
- Douglas, G.M.; Maffei, V.J.; Zaneveld, J.R.; Yurgel, S.N.; Brown, J.R.; Taylor, C.M.; Huttenhower, C.; Langille, M.G.I. PICRUSt2 for prediction of metagenome functions. Nat. Biotechnol. 2020, 38, 685–688. [Google Scholar] [CrossRef]
- Caspi, R.; Billington, R.; Fulcher, C.A.; Keseler, I.M.; Kothari, A.; Krummenacker, M.; Latendresse, M.; Midford, P.E.; Ong, Q.; Ong, W.K.; et al. The MetaCyc database of metabolic pathways and enzymes. Nucleic Acids Res. 2018, 46, D633–D639. [Google Scholar] [CrossRef] [Green Version]
- Parks, D.H.; Tyson, G.W.; Hugenholtz, P.; Beiko, R.G. STAMP: Statistical analysis of taxonomic and functional profiles. Bioinformatics 2014, 30, 3123–3124. [Google Scholar] [CrossRef] [Green Version]
- Thompson, F.L.; Gevers, D.; Thompson, C.C.; Dawyndt, P.; Naser, S.; Hoste, B.; Munn, C.B.; Swings, J. Phylogeny and molecular identification of vibrios on the basis of multilocus sequence analysis. Appl. Environ. Microbiol. 2005, 71, 5107–5115. [Google Scholar] [CrossRef] [Green Version]
- Valdes, A.M.; Walter, J.; Segal, E.; Spector, T.D. Role of the gut microbiota in nutrition and health. BMJ 2018, 361, k2179. [Google Scholar] [CrossRef] [Green Version]
- Ofek, T.; Lalzar, M.; Izhaki, I.; Halpern, M. Intestine and spleen microbiota composition in healthy and diseased tilapia. Anim. Microbiome 2022, 4, 50. [Google Scholar] [CrossRef]
- Llewellyn, M.S.; McGinnity, P.; Dionne, M.; Letourneau, J.; Thonier, F.; Carvalho, G.R.; Creer, S.; Derome, N. The biogeography of the atlantic salmon (Salmo salar) gut microbiome. ISME J. 2016, 10, 1280–1284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eilers, H.; Pernthaler, J.; Amann, R. Succession of Pelagic Marine Bacteria during Enrichment: A Close Look at Cultivation-Induced Shifts. Appl. Environ. Microbiol. 2000, 66, 4634–4640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thompson, F.L.; Iida, T.; Swings, J. Biodiversity of Vibrios. Microbiol. Mol. Biol. Rev. 2004, 68, 403–431. [Google Scholar] [CrossRef] [Green Version]
- Austin, B.; Austin, D.A. Aeromonadaceae representative (Aeromonas salmonicida). In Bacterial Fish Pathogens, Disease of Farmed and Wild Fish; Springer: Cham, Switzerland, 2016. [Google Scholar] [CrossRef]
- Mann, B.C.; Bezuidenhout, J.J.; Swanevelder, Z.H.; Grobler, A.F. MinION 16S datasets of a commercially available microbial community enables the evaluation of DNA extractions and data analyses. Data Brief 2021, 36, 107036. [Google Scholar] [CrossRef]
- Rozas, M.; Brillet, F.; Callewaert, C.; Paetzold, B. MinION Nanopore Sequencing of Skin Microbiome 16S and 16S-23S rRNA Gene Amplicons. Front. Cell Infect. Microbiol. 2021, 11, 806476. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Sham, R.C.; Deng, Y.; Mao, Y.; Wang, C.; Zhang, T.; Leung, K.M.Y. Diversity of gut microbiomes in marine fishes is shaped by host-related factors. Mol. Ecol. 2020, 29, 5019–5034. [Google Scholar] [CrossRef] [PubMed]
- Roques, S.; Deborde, C.; Richard, N.; Skiba-Cassy, S.; Moing, A.; Fauconneau, B. Metabolomics and fish nutrition: A review in the context of sustainable feed development. Rev. Aquac. 2018, 12, 261–282. [Google Scholar] [CrossRef] [Green Version]
- Díaz, E.; Ferrández, A.; Prieto, M.A.; García, J.L. Biodegradation of aromatic compounds by Escherichia coli. Microbiol. Mol. Biol. Rev. 2001, 65, 523–569, table of contents. [Google Scholar] [CrossRef] [Green Version]
- Rimoldi, S.; Torrecillas, S.; Montero, D.; Gini, E.; Makol, A.; Valdenegro, V.V.; Izquierdo, M.; Terova, G. Assessment of dietary supplementation with galactomannan oligosaccharides and phytogenics on gut microbiota of European sea bass (Dicentrarchus Labrax) fed low fishmeal and fish oil based diet. PLoS ONE 2020, 15, e0231494. [Google Scholar] [CrossRef]
- Dalsgaard, J.; Lund, I.; Thorarinsdottir, R.; Drengstig, A.; Arvonen, K.; Pedersen, P.B. Farming different species in RAS in Nordic countries: Current status and future perspectives. Aquac. Eng. 2013, 53, 2–13. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morales-Rivera, M.F.; Valenzuela-Miranda, D.; Nuñez-Acuña, G.; Benavente, B.P.; Gallardo-Escárate, C.; Valenzuela-Muñoz, V. Atlantic Salmon (Salmo salar) Transfer to Seawater by Gradual Salinity Changes Exhibited an Increase in The Intestinal Microbial Abundance and Richness. Microorganisms 2023, 11, 76. https://doi.org/10.3390/microorganisms11010076
Morales-Rivera MF, Valenzuela-Miranda D, Nuñez-Acuña G, Benavente BP, Gallardo-Escárate C, Valenzuela-Muñoz V. Atlantic Salmon (Salmo salar) Transfer to Seawater by Gradual Salinity Changes Exhibited an Increase in The Intestinal Microbial Abundance and Richness. Microorganisms. 2023; 11(1):76. https://doi.org/10.3390/microorganisms11010076
Chicago/Turabian StyleMorales-Rivera, María F., Diego Valenzuela-Miranda, Gustavo Nuñez-Acuña, Bárbara P. Benavente, Cristian Gallardo-Escárate, and Valentina Valenzuela-Muñoz. 2023. "Atlantic Salmon (Salmo salar) Transfer to Seawater by Gradual Salinity Changes Exhibited an Increase in The Intestinal Microbial Abundance and Richness" Microorganisms 11, no. 1: 76. https://doi.org/10.3390/microorganisms11010076