
Identification and Molecular Characterization of Four New Blastocystis Subtypes Designated ST35-ST38
 ,
,  , , , , , , , , and add
Show full author list
, , , , , , , , and add
Show full author list
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sources and Descriptions of Samples
2.2. PCR Amplification and Sequencing of the Full-Length ssu rRNA Gene
2.3. Bioinformatics Analysis (Read Processing and Consensus Building)
2.4. Phylogenetic Analyses
3. Results
3.1. Full-Length ssu rRNA Gene Sequences
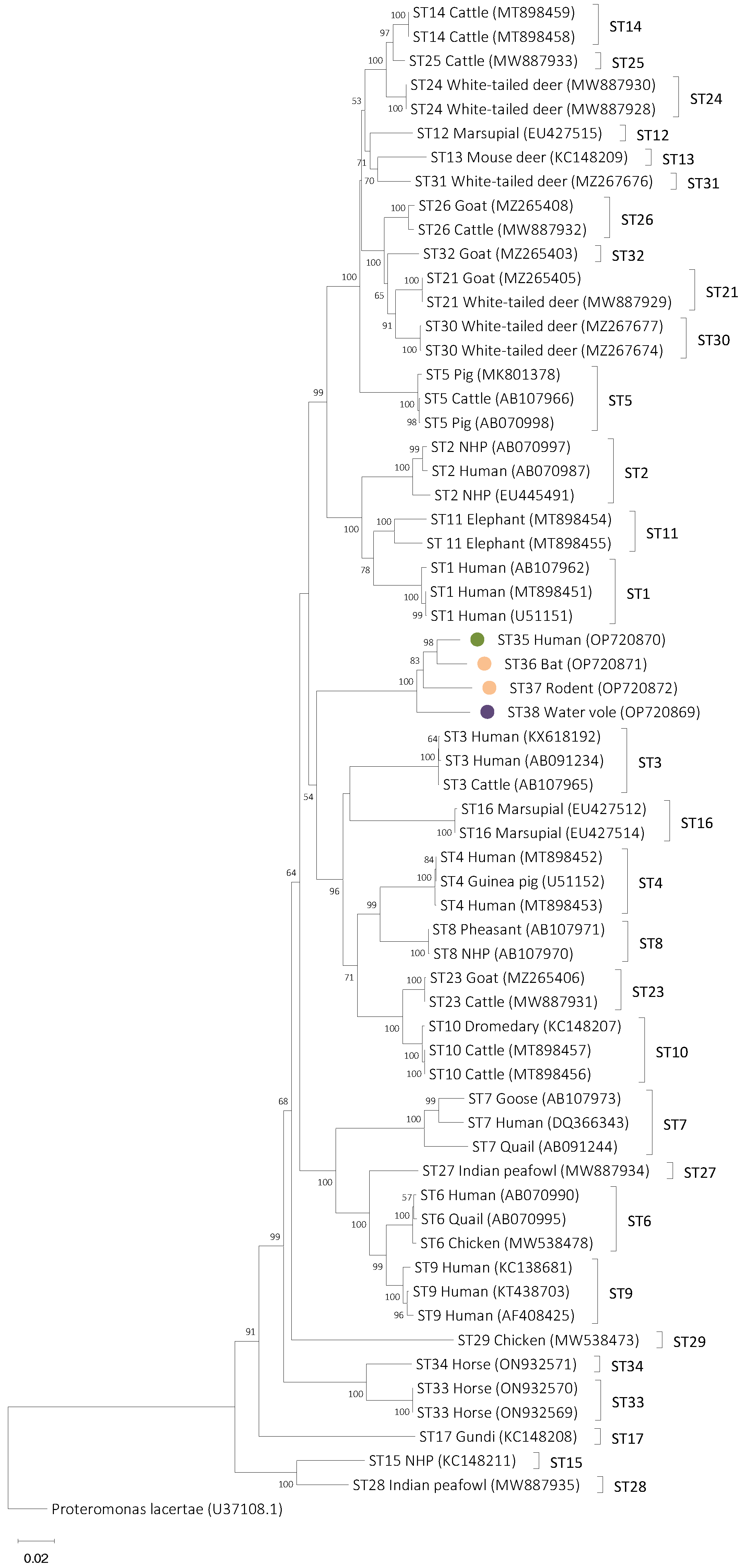
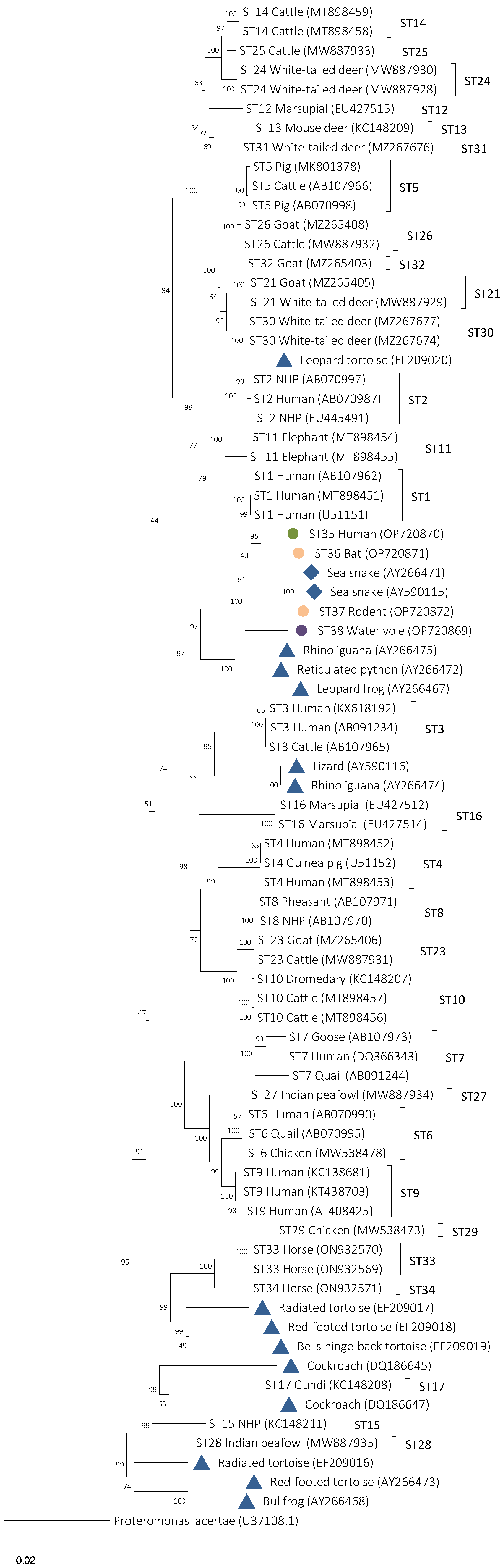
3.2. Phylogenetic Analyses
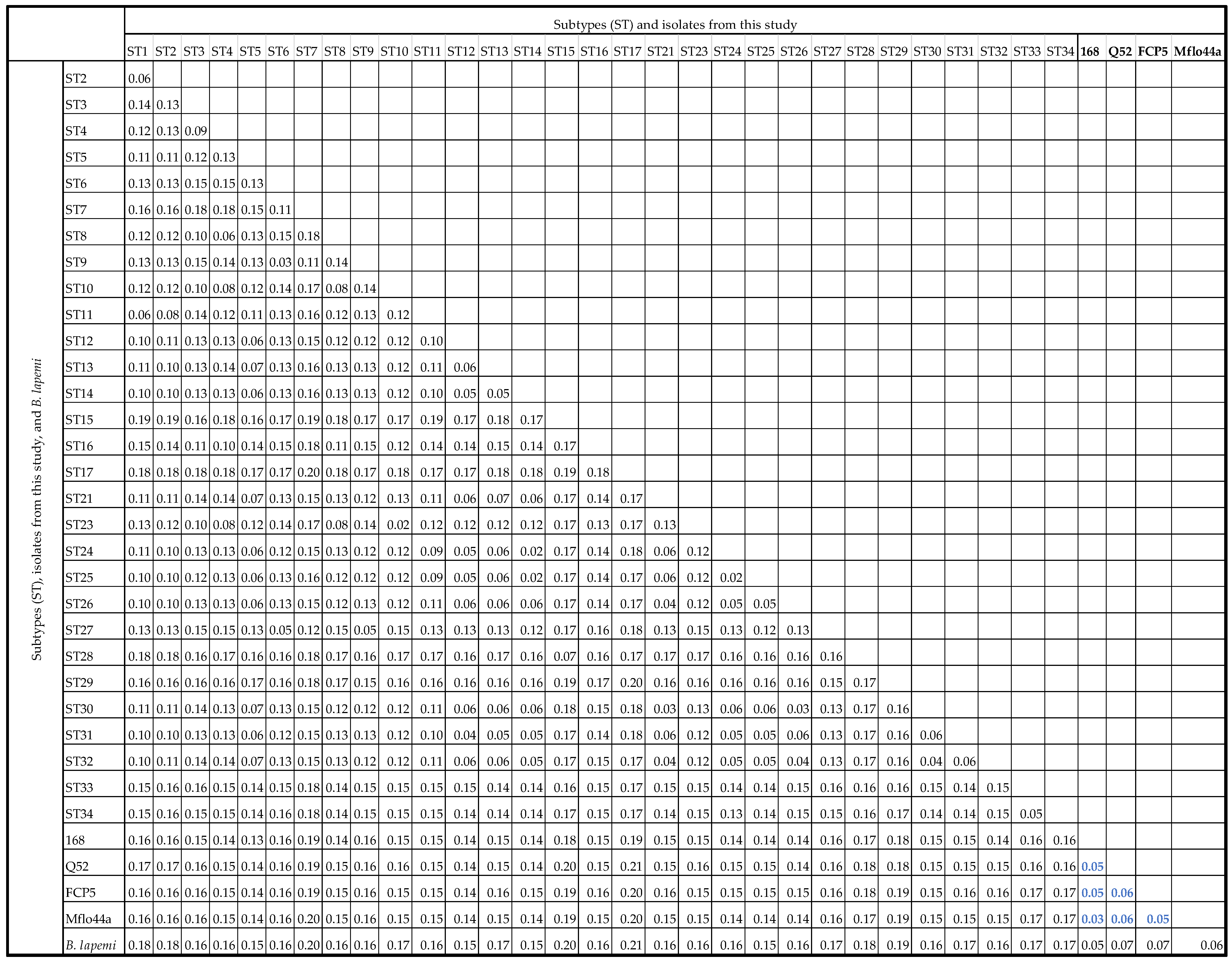
3.3. Pairwise Distance Comparisons
3.4. Designation of Blastocystis Subtypes ST35, ST36, ST37, and ST38
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Stensvold, C.R.; Clark, C.G. Pre-Empting Pandora’s Box: Blastocystis Subtypes Revisited. Trends Parasitol. 2020, 36, 229–232. [Google Scholar] [CrossRef] [PubMed]
- Hublin, J.S.Y.; Maloney, J.G.; Santin, M. Blastocystis in Domesticated and Wild Mammals and Birds. Res. Vet. Sci. 2021, 135, 260–282. [Google Scholar] [CrossRef] [PubMed]
- Tan, K.S.W. New Insights on Classification, Identification, and Clinical Relevance of Blastocystis spp. Clin. Microbiol. Rev. 2008, 21, 639–665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cifre, S.; Gozalbo, M.; Ortiz, V.; Soriano, J.M.; Merino, J.F.; Trelis, M. Blastocystis Subtypes and Their Association with Irritable Bowel Syndrome. Med. Hypotheses 2018, 116, 4–9. [Google Scholar] [CrossRef] [PubMed]
- Bahrami, F.; Babaei, E.; Badirzadeh, A.; Riabi, T.R.; Abdoli, A. Blastocystis, Urticaria, and Skin Disorders: Review of the Current Evidences. Eur. J. Clin. Microbiol. Infect. Dis. 2020, 39, 1027–1042. [Google Scholar] [CrossRef] [PubMed]
- Audebert, C.; Even, G.; Cian, A.; Blastocystis Investigation Group; Loywick, A.; Merlin, S.; Viscogliosi, E.; Chabé, M. Colonization with the Enteric Protozoa Blastocystis Is Associated with Increased Diversity of Human Gut Bacterial Microbiota. Sci. Rep. 2016, 6, 1–11. [Google Scholar] [CrossRef]
- Tito, R.Y.; Chaffron, S.; Caenepeel, C.; Lima-Mendez, G.; Wang, J.; Vieira-Silva, S.; Falony, G.; Hildebrand, F.; Darzi, Y.; Rymenans, L.; et al. Population-Level Analysis of Blastocystis Subtype Prevalence and Variation in the Human Gut Microbiota. Gut 2019, 68, 1180–1189. [Google Scholar] [CrossRef] [Green Version]
- Beghini, F.; Pasolli, E.; Truong, T.D.; Putignani, L.; Cacciò, S.M.; Segata, N. Large-Scale Comparative Metagenomics of Blastocystis, a Common Member of the Human Gut Microbiome. ISME J. 2017, 11, 2848–2863. [Google Scholar] [CrossRef]
- Stensvold, C.R.; Suresh, G.K.; Tan, K.S.W.; Thompson, R.C.A.; Traub, R.J.; Viscogliosi, E.; Yoshikawa, H.; Clark, C.G. Terminology for Blastocystis Subtypes—A Consensus. Trends Parasitol. 2007, 23, 93–96. [Google Scholar] [CrossRef]
- Baek, S.; Maloney, J.G.; Molokin, A.; George, N.S.; Cortés Vecino, J.A.; Santin, M. Diversity of Blastocystis Subtypes in Horses in Colombia and Identification of Two New Subtypes. Microorganisms 2022, 10, 1693. [Google Scholar] [CrossRef]
- Maloney, J.G.; Molokin, A.; Santin, M. Use of Oxford Nanopore MinION to Generate Full-Length Sequences of the Blastocystis Small Subunit (SSU) RRNA Gene. Parasites Vectors 2020, 13, 595. [Google Scholar] [CrossRef] [PubMed]
- Alfellani, M.A.; Stensvold, C.R.; Vidal-Lapiedra, A.; Onuoha, E.S.U.; Fagbenro-Beyioku, A.F.; Clark, C.G. Variable Geographic Distribution of Blastocystis Subtypes and Its Potential Implications. Acta Trop. 2013, 126, 11–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jinatham, V.; Maxamhud, S.; Popluechai, S.; Tsaousis, A.D.; Gentekaki, E. Blastocystis One Health Approach in a Rural Community of Northern Thailand: Prevalence, Subtypes and Novel Transmission Routes. Front. Microbiol. 2021, 12, 3800. [Google Scholar] [CrossRef] [PubMed]
- Khaled, S.; Gantois, N.; Ayoubi, A.; Even, G.; Sawant, M.; El Houmayraa, J.; Nabot, M.; Benamrouz-Vanneste, S.; Chabé, M.; Certad, G.; et al. Blastocystis sp. Prevalence and Subtypes Distribution amongst Syrian Refugee Communities Living in North Lebanon. Microorganisms 2021, 9, 184. [Google Scholar] [CrossRef] [PubMed]
- Khaled, S.; Gantois, N.; Ly, A.T.; Senghor, S.; Even, G.; Dautel, E.; Dejager, R.; Sawant, M.; Baydoun, M.; Benamrouz-Vanneste, S.; et al. Prevalence and Subtype Distribution of Blastocystis sp. in Senegalese School Children. Microorganisms 2020, 8, 408. [Google Scholar] [CrossRef]
- Osorio-Pulgarin, M.I.; Higuera, A.; Beltran-álzate, J.C.; Sánchez-Jiménez, M.; Ramírez, J.D. Epidemiological and Molecular Characterization of Blastocystis Infection in Children Attending Daycare Centers in Medellín, Colombia. Biology 2021, 10, 669. [Google Scholar] [CrossRef]
- Ramírez, J.D.; Sánchez, A.; Hernández, C.; Flórez, C.; Bernal, M.C.; Giraldo, J.C.; Reyes, P.; López, M.C.; García, L.; Cooper, P.J.; et al. Geographic Distribution of Human Blastocystis Subtypes in South America. Infect. Genet. Evol. 2016, 41, 32–35. [Google Scholar] [CrossRef]
- Seguí, R.; Muñoz-Antoli, C.; Klisiowicz, D.R.; Oishi, C.Y.; Köster, P.C.; de Lucio, A.; Hernández-de-Mingo, M.; Puente, P.; Toledo, R.; Esteban, J.G.; et al. Prevalence of Intestinal Parasites, with Emphasis on the Molecular Epidemiology of Giardia duodenalis and Blastocystis sp., in the Paranaguá Bay, Brazil: A Community Survey. Parasites Vectors 2018, 11, 490. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Hernandez, F.; Martinez-Ibarra, J.A.; Lopez-Escamilla, E.; Villanueva-Garcia, C.; Muñoz-Garcia, C.I.; Rendon-Franco, E.; Maravilla, P.; Villalobos, G. Molecular Genotyping of Blastocystis spp. in Wild Mammals from Mexico. Parasitol. Res. 2020, 119, 97–104. [Google Scholar] [CrossRef]
- Betts, E.L.; Gentekaki, E.; Tsaousis, A.D. Exploring Micro-Eukaryotic Diversity in the Gut: Co-Occurrence of Blastocystis Subtypes and Other Protists in Zoo Animals. Front. Microbiol. 2020, 11, 288. [Google Scholar] [CrossRef]
- Teow, W.L.; Zaman, V.; Ng, G.C.; Chan, Y.C.; Yap, E.H.; Howe, J.; Gopalakrishnakone, P.; Singh, M. A Blastocystis Species from the Sea-Snake, Lapemis Hardwickii (Serpentes: Hydrophiidae). Int. J. Parasitol. 1991, 21, 723–726. [Google Scholar] [CrossRef] [PubMed]
- Betts, E.L.; Hoque, S.; Torbe, L.; Bailey, J.R.; Ryan, H.; Toller, K.; Breakell, V.; Carpenter, A.I.; Diana, A.; Matechou, E.; et al. Parasites, Drugs and Captivity: Blastocystis-Microbiome Associations in Captive Water Voles. Biology 2021, 10, 457. [Google Scholar] [CrossRef] [PubMed]
- Rognes, T.; Flouri, T.; Nichols, B.; Quince, C.; Mahé, F. VSEARCH: A versatile open source tool for metagenomics. PeerJ 2016, 4, e2584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K.; Battistuzzi, F.U. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Maloney, J.G.; Santin, M. Mind the Gap: New Full-Length Sequences of Blastocystis Subtypes Generated via Oxford Nanopore Minion Sequencing Allow for Comparisons between Full-Length and Partial Sequences of the Small Subunit of the Ribosomal RNA Gene. Microorganisms 2021, 9, 997. [Google Scholar] [CrossRef]
- Higuera, A.; Herrera, G.; Jimenez, P.; García-Corredor, D.; Pulido-Medellín, M.; Bulla-Castañeda, D.M.; Pinilla, J.C.; Moreno-Pérez, D.A.; Maloney, J.G.; Santín, M.; et al. Identification of Multiple Blastocystis Subtypes in Domestic Animals From Colombia Using Amplicon-Based Next Generation Sequencing. Front. Vet. Sci. 2021, 8, 932. [Google Scholar] [CrossRef]
- Maloney, J.G.; Jang, Y.; Molokin, A.; George, N.S.; Santin, M. Wide Genetic Diversity of Blastocystis in White-Tailed Deer (Odocoileus Virginianus) from Maryland, USA. Microorganisms 2021, 9, 1343. [Google Scholar] [CrossRef]
- Yoshikawa, H.; Morimoto, K.; Wu, Z.; Singh, M.; Hashimoto, T. Problems in Speciation in the Genus Blastocystis. Trends Parasitol. 2004, 20, 251–255. [Google Scholar] [CrossRef]
- Yoshikawa, H.; Koyama, Y.; Tsuchiya, E.; Takami, K. Blastocystis Phylogeny among Various Isolates from Humans to Insects. Parasitol. Int. 2016, 65, 750–759. [Google Scholar] [CrossRef]
- Cian, A.; El Safadi, D.; Osman, M.; Moriniere, R.; Gantois, N.; Benamrouz-Vanneste, S.; Delgado-Viscogliosi, P.; Guyot, K.; Li, L.L.; Monchy, S.; et al. Molecular Epidemiology of Blastocystis sp. in Various Animal Groups from Two French Zoos and Evaluation of Potential Zoonotic Risk. PLoS ONE 2017, 12, e0169659. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maloney, J.G.; Molokin, A.; Seguí, R.; Maravilla, P.; Martínez-Hernández, F.; Villalobos, G.; Tsaousis, A.D.; Gentekaki, E.; Muñoz-Antolí, C.; Klisiowicz, D.R.; et al. Identification and Molecular Characterization of Four New Blastocystis Subtypes Designated ST35-ST38. Microorganisms 2023, 11, 46. https://doi.org/10.3390/microorganisms11010046
Maloney JG, Molokin A, Seguí R, Maravilla P, Martínez-Hernández F, Villalobos G, Tsaousis AD, Gentekaki E, Muñoz-Antolí C, Klisiowicz DR, et al. Identification and Molecular Characterization of Four New Blastocystis Subtypes Designated ST35-ST38. Microorganisms. 2023; 11(1):46. https://doi.org/10.3390/microorganisms11010046
Chicago/Turabian StyleMaloney, Jenny G., Aleksey Molokin, Raimundo Seguí, Pablo Maravilla, Fernando Martínez-Hernández, Guiehdani Villalobos, Anastasios D. Tsaousis, Eleni Gentekaki, Carla Muñoz-Antolí, Debora R. Klisiowicz, and et al. 2023. "Identification and Molecular Characterization of Four New Blastocystis Subtypes Designated ST35-ST38" Microorganisms 11, no. 1: 46. https://doi.org/10.3390/microorganisms11010046