
Genome Characterization of Bacteriophage KPP-1, a Novel Member in the Subfamily Vequintavirinae, and Use of Its Endolysin for the Lysis of Multidrug-Resistant Klebsiella variicola In Vitro

Abstract
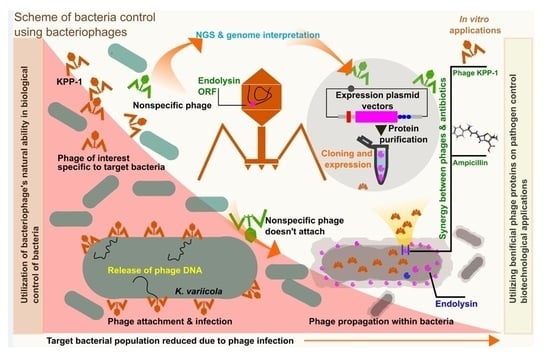
:
1. Introduction
2. Materials and Methods
2.1. Bacterial Strains and Culture Conditions
2.2. Isolation of Bacteriophage KPP-1 That Infects K. variicola
2.3. High Titer Bacteriophage Preparation
2.4. Host Range Analysis
2.5. Transmission Electron Microscopy (TEM)
2.6. Genomic DNA Isolation and KPP-1 Phage Genome Sequencing
2.7. Functional Annotation
2.8. Comparative Genomic Analysis and Phylogenetic Positioning
2.9. Synergistic Inhibition of Bacterial Growth by KPP-1 and Antibiotics
2.10. Identification and Cloning of Endolysin
2.11. Determination of the Effect of KPP-1 Endolysin on Bacterial Cells Using Live and Dead Cell Assay
2.12. Effect of KPP-1 Endolysin on Bacterial Cells upon Oxidative Stress
2.13. Antibacterial Activity of KPP-1 Endolysin
2.14. Statistical Analysis
3. Results
3.1. Isolation, Morphology, Propagation Host, and the Host Range of KPP-1
3.2. Complete Genome Sequencing and Functional Annotation of KPP-1
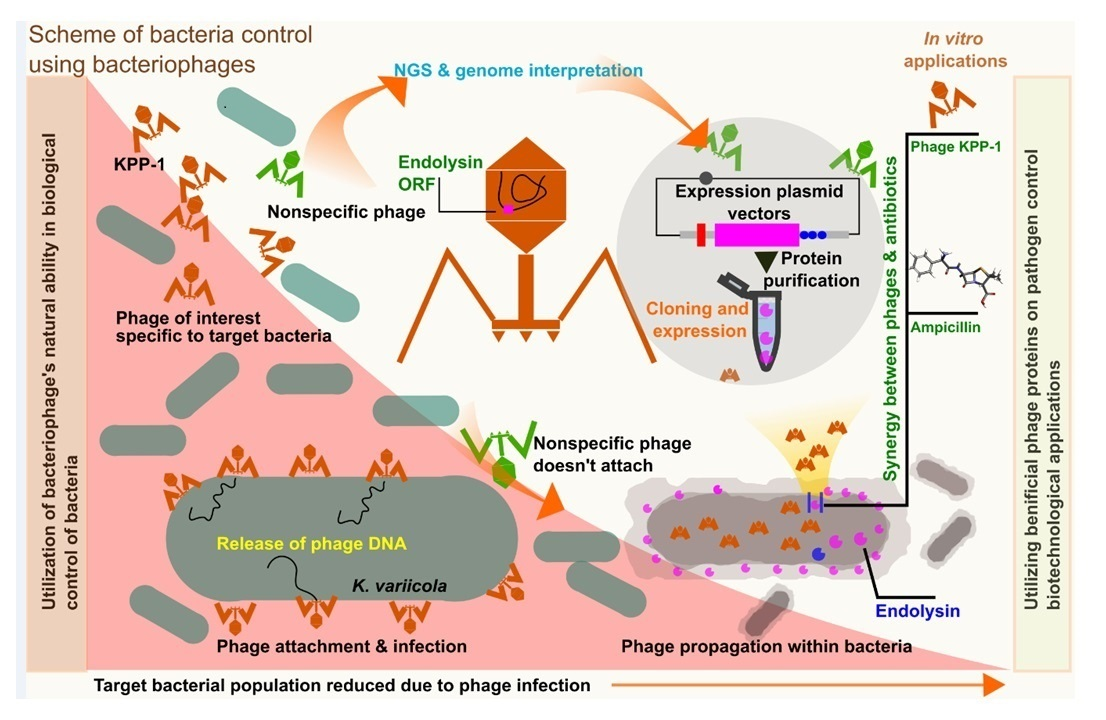
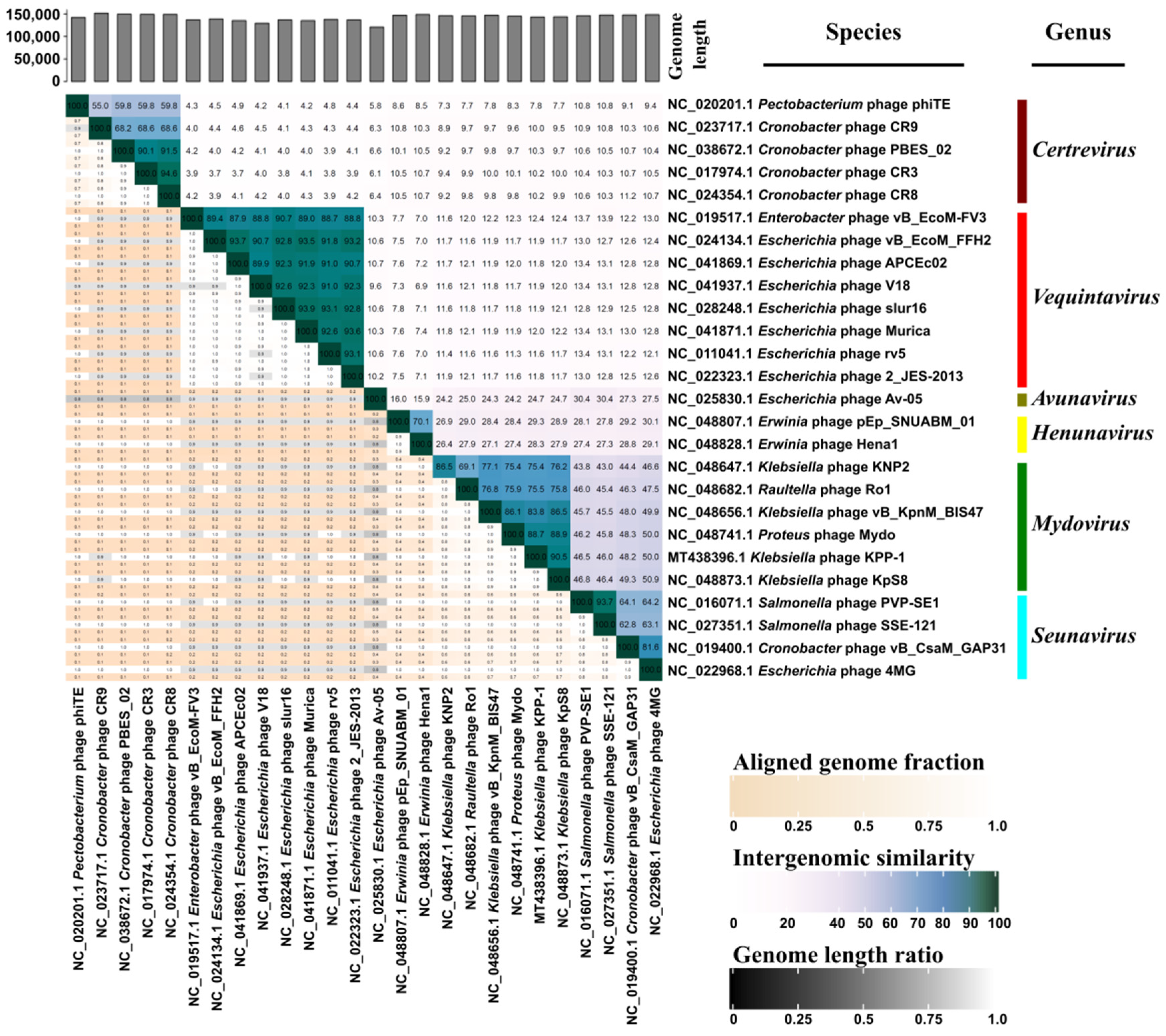
3.3. Comparative Genomic Analysis and Phylogenetic Classification
3.4. Bacterial Growth Inhibition by KPP-1 and Its Synergy with Antibiotics
3.5. Identification of the Endolysin Gene in the KPP-1 Genome
3.6. The Effect of KPP-1 Endolysin on the Bacterial Cell Membrane
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Rodríguez-Medina, N.; Barrios-Camacho, H.; Duran-Bedolla, J.; Garza-Ramos, U. Klebsiella variicola: An Emerging Pathogen in Humans. Emerg. Microbes Infect. 2019, 8, 973–988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brisse, S.; Passet, V.; Grimont, P.A.D. Description of Klebsiella quasipneumoniae sp. nov., Isolated from Human Infections, With Two Subspecies, Klebsiella quasipneumoniae subsp. quasipneumoniae subsp. nov. and Klebsiella quasipneumoniae subsp. similipneumoniae subsp. nov., and Demonstration that Klebsiella Singaporeans is Junior Heterotypic Synonym of Klebsiella variicola. Int. J. Syst. Evol. Microbiol. 2014, 64, 3146–3152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brisse, S.; Verhoef, J. Phylogenetic Diversity of Klebsiella pneumoniae and Klebsiella oxytoca Clinical Isolates Revealed by Randomly Amplified Polymorphic DNA, gyrA and parC Genes Sequencing and Automated Rribotyping. Int. J. Syst. Evol. Microbiol. 2001, 51, 915–924. [Google Scholar] [CrossRef]
- Wang, Y.; Zhu, B.; Liu, M.; Dong, X.; Ma, J.; Li, X.; Cheng, F.; Guo, J.; Lu, S.; Wan, F.; et al. Characterization of IncHI1B Plasmids Encoding Efflux Pump TmexCD2-ToprJ2 in Carbapenem-Resistant Klebsiella variicola, Klebsiella quasipneumoniae, and Klebsiella michiganensis Strains. Front. Microbiol. 2021, 12, 759208. [Google Scholar] [CrossRef] [PubMed]
- Berry, G.J.; Loeffelholz, M.J.; Williams-Bouyer, N. An Investigation into Laboratory Misidentification of a Bloodstream Klebsiella variicola Infection. J. Clin. Microbiol. 2015, 53, 2793–2794. [Google Scholar] [CrossRef] [Green Version]
- Maatallah, M.; Vading, M.; Kabir, M.H.; Bakhrouf, A.; Kalin, M.; Naucler, P.; Brisse, S.; Giske, C.G. Klebsiella variicola Is a Frequent Cause of Bloodstream Infection in the Stockholm Area and Associated with Higher Mortality Compared to K. pneumoniae. PLoS ONE 2014, 9, e113539. [Google Scholar] [CrossRef] [Green Version]
- Garza-Ramos, U.; Moreno-Dominguez, S.; Hernández-Castro, R.; Silva-Sanchez, J.; Barrios, H.; Reyna-Flores, F.; Sanchez-Perez, A.; Carrillo-Casas, E.M.; Sanchez-León, M.C.; Moncada-Barron, D. Identification and Characterization of Imipenem-Resistant Klebsiella pneumoniae and Susceptible Klebsiella variicola Isolates Obtained from the Same Patient. Microb. Drug Resist. 2016, 22, 179–184. [Google Scholar] [CrossRef] [PubMed]
- Kiley, J.L.; Mende, K.; Beckius, M.L.; Kaiser, S.J.; Carson, M.L.; Lu, D.; Whitman, T.J.; Petfield, J.L.; Tribble, D.R.; Blyth, D.M. Resistance Patterns and Clinical Outcomes of Klebsiella pneumoniae and Invasive Klebsiella variicola in Trauma Patients. PLoS ONE 2021, 16, e0255636. [Google Scholar] [CrossRef]
- Akine, D.; Sasahara, T.; Watanabe, S.; Ishishita, Y.; Yamaguchi, T.; Cui, L.; Morisawa, Y. Post-Surgical Meningitis Caused by Klebsiella variicola. IDCases 2019, 18, e00622. [Google Scholar] [CrossRef] [PubMed]
- Alfaleh, M.A.; Alsaab, H.O.; Mahmoud, A.B.; Alkayyal, A.A.; Jones, M.L.; Mahler, S.M.; Hashem, A.M. Phage Display Derived Monoclonal Antibodies: From Bench to Bedside. Front. Immunol. 2020, 11, 1986. [Google Scholar] [CrossRef]
- Zhang, W.; Wu, Q. Applications of Phage-Derived RNA-Based Technologies in Synthetic Biology. Synth. Syst. Biotechnol. 2020, 5, 343–360. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, L.; Cima-Cabal, M.D.; Duarte, A.C.; Rodríguez, A.; García-Suárez, M.D.M.; García, P. Gram-Positive Pneumonia: Possibilities Offered by Phage Therapy. Antibiotics 2021, 10, 1000. [Google Scholar] [CrossRef] [PubMed]
- Qadir, M.I.; Sajjad, S. Phage Therapy Against Streptococcus pneumoniae: Modern Tool to Control Pneumonia. Crit. Rev. Eukaryot. Gene Expr. 2017, 27, 289–295. [Google Scholar] [CrossRef] [PubMed]
- Guerrero-Bustamante, C.A.; Dedrick, R.M.; Garlena, R.A.; Russell, D.A.; Hatfull, G.F. Toward a Phage Cocktail for tuberculosis: Susceptibility and Tuberculocidal Action of Mycobacteriophages Against Diverse Mycobacterium tuberculosis Strains. MBio 2021, 12, e00973-21. [Google Scholar] [CrossRef] [PubMed]
- Guo, M.; Gao, Y.; Xue, Y.; Liu, Y.; Zeng, X.; Cheng, Y.; Ma, J.; Wang, H.; Sun, J.; Wang, Z.; et al. Bacteriophage Cocktails Protect Dairy Cows Against Mastitis Caused by Drug Resistant Escherichia coli Infection. Front. Cell. Infect. Microbiol. 2021, 11, 690377. [Google Scholar] [CrossRef]
- Nakai, T.; Park, S.C. Bacteriophage Therapy of Infectious Diseases in Aquaculture. Res. Microbiol. 2002, 153, 13–18. [Google Scholar] [CrossRef]
- Schoch, C.L.; Ciufo, S.; Domrachev, M.; Hotton, C.L.; Kannan, S.; Khovanskaya, R.; Leipe, D.; McVeigh, R.; O’Neill, K.; Robbertse, B.; et al. NCBI Taxonomy: A Comprehensive Update on Curation, Resources and Tools. Database 2020, 2020, baaa062. [Google Scholar] [CrossRef]
- Chang, Y. Bacteriophage-derived Endolysins Applied as Potent Biocontrol Agents to Enhance Food Safety. Microorganisms 2020, 8, 724. [Google Scholar] [CrossRef]
- Tolba, M.; Ahmed, M.U.; Tlili, C.; Eichenseher, F.; Loessner, M.J.; Zourob, M. A Bacteriophage Endolysin-Based Electrochemical Impedance Biosensor for the Rapid Detection of Listeria Cells. Analyst 2012, 137, 5749–5756. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic Local Alignment Search Tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Kim, O.S.; Cho, Y.J.; Lee, K.; Yoon, S.H.; Kim, M.; Na, H.; Park, S.C.; Jeon, Y.S.; Lee, J.H.; Yi, H.; et al. Introducing EzTaxon-e: A Prokaryotic 16s rRNA Gene Sequence Database with Phylotypes that Represent Uncultured Species. Int. J. Syst. Evol. Microbiol. 2012, 62, 716–721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, H.; Bandara, N.; Shin, E.; Ryu, S.; Kim, K. Pyo Prevalence of Bacillus cereus Bacteriophages in Fermented Foods and Characterization of Phage JBP901. Res. Microbiol. 2011, 162, 791–797. [Google Scholar] [CrossRef]
- Rai, S.; Tyagi, A.; Kumar, B.T.N.; Singh, N.K. Optimization of Plaque Forming Conditions for an Aeromonas hydrophila Lytic Bacteriophage. Int. J. Curr. Microbiol. Appl. Sci. 2020, 9, 3764–3768. [Google Scholar] [CrossRef]
- Ghosh, K.; Senevirathne, A.; Kang, H.S.; Hyun, W.B.; Kim, J.E.; Kim, K.P. Complete Nucleotide Sequence Analysis of a Novel Bacillus subtilis-Infecting Bacteriophage BSP10 and its Effect on Poly-Gamma-Glutamic Acid Degradation. Viruses 2018, 10, 240. [Google Scholar] [CrossRef] [Green Version]
- Bandara, N.; Jo, J.; Ryu, S.; Kim, K.P. Bacteriophages BCP1-1 and BCP8-2 Require Divalent Cations for Efficient Control of Bacillus cereus in Fermented Foods. Food Microbiol. 2012, 31, 9–16. [Google Scholar] [CrossRef]
- Rueden, C.T.; Schindelin, J.; Hiner, M.C.; DeZonia, B.E.; Walter, A.E.; Arena, E.T.; Eliceiri, K.W. ImageJ2: ImageJ for the Next Generation of Scientific Image Data. BMC Bioinform. 2017, 18, 529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, A.D.; de Sena Brandine, G. Falco: High-speed FastQC Emulation for Quality Control of Sequencing Data. F1000Research 2021, 8, 1874. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A New Genome Assembly Algorithm and Its Applications to Single-Cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [Green Version]
- Seemann, T. Prokka: Rapid Prokaryotic Genome Annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef] [Green Version]
- Aziz, R.K.; Bartels, D.; Best, A.; DeJongh, M.; Disz, T.; Edwards, R.A.; Formsma, K.; Gerdes, S.; Glass, E.M.; Kubal, M.; et al. The RAST Server: Rapid Annotations Using Subsystems Technology. BMC Genom. 2008, 9, 75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delcher, A.L.; Bratke, K.A.; Powers, E.C.; Salzberg, S.L. Identifying Bacterial Genes and Endosymbiont DNA with Glimmer. Bioinformatics 2007, 23, 673–679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lomsadze, A.; Bonny, C.; Strozzi, F.; Borodovsky, M. GeneMark-HM: Improving Gene Prediction in DNA Sequences of Human Microbiome. NAR Genom. Bioinform. 2021, 3, lqab047. [Google Scholar] [CrossRef]
- Alvi, Z.A.; Chu, T.-C.; Schawaroch, V.; Klaus, A.V. Genomic and Expression Analysis of Transition Proteins in Drosophila. Spermatogenesis 2015, 5, e1178518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laslett, D.; Canback, B. ARAGORN, A Program to Detect tRNA Genes and mRNA Genes in Nucleotide Sequences. Nucleic Acids Res. 2004, 32, 11–16. [Google Scholar] [CrossRef]
- Chan, P.P.; Lowe, T.M. tRNAscan-SE: Searching for tRNA Genes in Genomic Sequences. In Methods in Molecular Biology; Humana: New York, NY, USA, 2019; Volume 1962, pp. 1–14. [Google Scholar] [CrossRef]
- Grant, J.R.; Stothard, P. The CGView Server: A Comparative Genomics Tool for Circular Genomes. Nucleic Acids Res. 2008, 36, W181–W184. [Google Scholar] [CrossRef]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An Integrated and Extendable Desktop Software Platform for the Organization and Analysis of Sequence data. Bioinformatics 2012, 12, 1647–1649. [Google Scholar] [CrossRef] [Green Version]
- Garneau, J.R.; Depardieu, F.; Fortier, L.C.; Bikard, D.; Monot, M. PhageTerm: A Tool for Fast and Accurate Determination of Phage Termini and Packaging Mechanism using Next-Generation Sequencing Data. Sci. Rep. 2017, 7, 8292. [Google Scholar] [CrossRef]
- International Committee on Taxonomy of Viruses. Available online: http://www.ictvonline.org/virusTaxonomy.asp (accessed on 3 March 2017).
- Moraru, C.; Varsani, A.; Kropinski, A.M. VIRIDIC—A Novel Tool to Calculate the Intergenomic Similarities of Prokaryote-Infecting Viruses. Viruses 2020, 12, 1268. [Google Scholar] [CrossRef]
- Sullivan, M.J.; Petty, N.K.; Beatson, S.A. Easyfig: A Genome Comparison Visualizer. Bioinformatics 2011, 27, 1009–1010. [Google Scholar] [CrossRef] [Green Version]
- Frickey, T.; Lupas, A. CLANS: A Java Application for Visualizing Protein Families Based on Pairwise Similarity. Bioinformatics 2004, 20, 3702–3704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dereeper, A.; Guignon, V.; Blanc, G.; Audic, S.; Buffet, S.; Chevenet, F.; Dufayard, J.F.; Guindon, S.; Lefort, V.; Lescot, M.; et al. Phylogeny.fr: Robust phylogenetic analysis for the non-specialist. Nucleic Acids Res. 2008, 36, W456–W459. [Google Scholar] [CrossRef]
- Rohwer, F.; Edwards, R. The Phage Proteomic Tree: A Genome-Based Taxonomy for Phage. J. Bacteriol. 2002, 184, 4529–4535. [Google Scholar] [CrossRef] [Green Version]
- Spriestersbach, A.; Kubicek, J.; Schäfer, F.; Block, H.; Maertens, B. Purification of His-Tagged Proteins. In Methods in Enzymology; Academic Press: Cambridge, MA, USA, 2015; Volume 559, pp. 1–15. [Google Scholar] [CrossRef]
- Kruger, N.J. The Bradford Method for Protein Quantitation. In The Protein Protocols Handbook; Springer: Manhattan, NY, USA, 1996; ISBN 978-1-59259-169-5. [Google Scholar]
- Biasini, M.; Bienert, S.; Waterhouse, A.; Arnold, K.; Studer, G.; Schmidt, T.; Kiefer, F.; Cassarino, T.G.; Bertoni, M.; Bordoli, L. SWISS-MODEL: Modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res. 2014, 42, W252–W258. [Google Scholar] [CrossRef]
- Quevillon, E.; Silventoinen, V.; Pillai, S.; Harte, N.; Mulder, N.; Apweiler, R.; Lopez, R. InterProScan: Protein domains identifier. Nucleic Acids Res. 2005, 33, W116–W120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yerushalmy, O.; Coppenhagen-Glazer, S.; Nir-Paz, R.; Tuomala, H.; Skurnik, M.; Kiljunen, S.; Hazan, R. Complete Genome Sequences of Two Klebsiella pneumoniae Phages Isolated as Part of an International Effort. Microbiol. Resour. Announc. 2019, 8, e00843-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thurgood, T.L.; Sharma, R.; Call, J.J.; Chronis, J.D.; Dawson, D.D.; Finnegan, Z.K.; Foster, K.W.; Meek, T.; Potts, E.; Sirrine, M.R.; et al. Genome Sequences of 12 Phages That Infect Klebsiella pneumoniae. Microbiol. Resour. Announc. 2020, 9, e00024-20. [Google Scholar] [CrossRef] [Green Version]
- Current Taxonomic Release. Available online: https://ictv.global/taxonomy (accessed on 12 October 2022).
- Marques, A.T.; Tanoeiro, L.; Duarte, A.; Gonçalves, L.; Vítor, J.M.B.; Vale, F.F. Genomic Analysis of Prophages from Klebsiella pneumoniae Clinical Isolates. Microorganisms 2021, 9, 2252. [Google Scholar] [CrossRef]
- Gasteiger, E.; Hoogland, C.; Gattiker, A.; Duvaud, S.; Wilkins, M.R.; Appel, R.D.; Bairoch, A. Protein Identification and Analysis Tools on the ExPASy Server. In The Proteomics Protocols Handbook; Humana: New York, NY, USA, 2005; Volume 112, pp. 531–552. [Google Scholar]
- Kanade, S.; Nataraj, G.; Ubale, M.; Mehta, P. Fluorescein Diacetate Vital Staining for Detecting Viability of Acid-Fast Bacilli in Patients on Antituberculosis Treatment. Int. J. Mycobacteriol. 2016, 5, 294–298. [Google Scholar] [CrossRef] [Green Version]
- Farr, S.B.; Kogoma, T. Oxidative Stress Responses in Escherichia coli and Salmonella Typhimurium. Microbiol. Rev. 1991, 55, 561–585. [Google Scholar] [CrossRef]
- Ackermann, H.W. Phage Classification and Characterization. Methods Mol. Biol. 2009, 501, 124–140. [Google Scholar] [CrossRef]
- Turner, D.; Kropinski, A.M.; Adriaenssens, E.M. A Roadmap for Genome-Based Phage Taxonomy. Viruses 2021, 13, 506. [Google Scholar] [CrossRef] [PubMed]
- Redrejo-Rodríguez, M.; Salas, M. Multiple Roles of Genome-Attached Bacteriophage Terminal Proteins. Virology 2014, 468–470, 322–329. [Google Scholar] [CrossRef] [Green Version]
- Casjens, S.R.; Gilcrease, E.B. Determining DNA Packaging Strategy by Analysis of the Termini of the Chromosomes in Tailed-Bacteriophage Virions. Methods Mol. Biol. 2009, 502, 91–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moghadam, M.T.; Khoshbayan, A.; Chegini, Z.; Farahani, I.; Shariati, A. Bacteriophages, a New Therapeutic Solution for Inhibiting Multidrug-Resistant Bacteria Causing Wound Infection: Lesson From Animal Models and Clinical Trials. Drug Des. Devel. Ther. 2020, 14, 1867–1883. [Google Scholar] [CrossRef]
- Akilandeswari, K.; Ruckmani, K. Synergistic Antibacterial Effect of Apigenin with ß-Lactam Antibiotics and Modulation of Bacterial Resistance by a Possible Membrane Effect Against Methicillin Resistant Staphylococcus aureus. Cell. Mol. Biol. 2016, 62, 74–82. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.; Kim, J.; Son, B.; Ryu, S. Development of Advanced Chimeric Endolysin to Control Multidrug-Resistant Staphylococcus aureus through Domain Shuffling. ACS Infect. Dis. 2021, 7, 2081–2092. [Google Scholar] [CrossRef]
- Young, R.; Bläsi, U. Holins: Form and Function in Bacteriophage Lysis. FEMS Microbiol. Rev. 1995, 17, 191–205. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bacteriophage | Genbank Accession No. | Genome Length (Kb) | G+C mol% | No. of CDS | No. of tRNAs | Genes | Pseudogenes | Query Cover (A) | % Identity (B) | Sequence Identity (A × B) |
|---|---|---|---|---|---|---|---|---|---|---|
| Klebsiella phage KPP-1 | MT438396.1 | 143.37 | 44.7 | 255 | 17 | 272 | 1 | 100 | 100.00 | 100.00 |
| Klebsiella phage vB_KpnM_KB57 | NC_028659.1 | 142.99 | 44.6 | 245 | 16 | 261 | 1 | 92 | 98.06 | 90.21 |
| Klebsiella phage vB_KpnM_BIS47 | NC_048656.1 | 147.44 | 44.6 | 262 | 0 | 262 | 0 | 84 | 96.16 | 80.77 |
| Proteus phage Mydo | NC_048741.1 | 145.13 | 44.8 | 264 | 23 | 287 | 0 | 89 | 98.80 | 87.93 |
| Mydovirus KpS8 | NC_048873.1 | 143.8 | 44.6 | 261 | 17 | 278 | 0 | 91 | 99.55 | 90.59 |
| Klebsiella phage KNP2 | NC_048647.1 | 146.2 | 45.2 | 211 | 0 | 211 | 0 | 76 | 98.62 | 74.95 |
| Salmonella phage PVPSE1 | NC_016071.1 | 145.96 | 45.6 | 244 | 24 | 268 | 0 | 25 | 77.73 | 19.43 |
| Salmonella phage SSE121 | NC_027351.1 | 147.74 | 45.3 | 242 | 0 | 242 | 0 | 26 | 77.73 | 20.20 |
| Erwinia phage Hena1 | NC_048828.1 | 148.84 | 48.4 | 240 | 26 | 266 | 0 | 8 | 78.01 | 6.24 |
| Erwinia phage pEp_SNUABM_01 | NC_048807.1 | 147.32 | 48.7 | 249 | 26 | 275 | 0 | 8 | 80.38 | 6.43 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Senevirathne, A.; Lee, J.; De Zoysa, M.; Nikapitiya, C. Genome Characterization of Bacteriophage KPP-1, a Novel Member in the Subfamily Vequintavirinae, and Use of Its Endolysin for the Lysis of Multidrug-Resistant Klebsiella variicola In Vitro. Microorganisms 2023, 11, 207. https://doi.org/10.3390/microorganisms11010207
Senevirathne A, Lee J, De Zoysa M, Nikapitiya C. Genome Characterization of Bacteriophage KPP-1, a Novel Member in the Subfamily Vequintavirinae, and Use of Its Endolysin for the Lysis of Multidrug-Resistant Klebsiella variicola In Vitro. Microorganisms. 2023; 11(1):207. https://doi.org/10.3390/microorganisms11010207
Chicago/Turabian StyleSenevirathne, Amal, Jehee Lee, Mahanama De Zoysa, and Chamilani Nikapitiya. 2023. "Genome Characterization of Bacteriophage KPP-1, a Novel Member in the Subfamily Vequintavirinae, and Use of Its Endolysin for the Lysis of Multidrug-Resistant Klebsiella variicola In Vitro" Microorganisms 11, no. 1: 207. https://doi.org/10.3390/microorganisms11010207