
Effect of Climate on Bacterial and Archaeal Diversity of Moroccan Marine Microbiota

 ,
,  , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Selection and Description of Sampling Sites
2.2. Sampling
2.3. Sample Preparation and DNA Extraction
2.4. PCR Amplification
2.5. Illumina Sequencing and Data Analysis
2.5.1. 16S rRNA Gene Sequencing
2.5.2. Sequence Analysis
2.5.3. Taxonomic Profiling of the Data
2.5.4. Functional Profiling
2.5.5. Comparative Analysis
2.5.6. Statistical Analysis
2.6. Sequence Availability
3. Results
3.1. Physical and Chemical Parameters of Water Samples
3.2. Taxonomic Composition Based on Total Community
3.3. Alpha and Beta Diversity
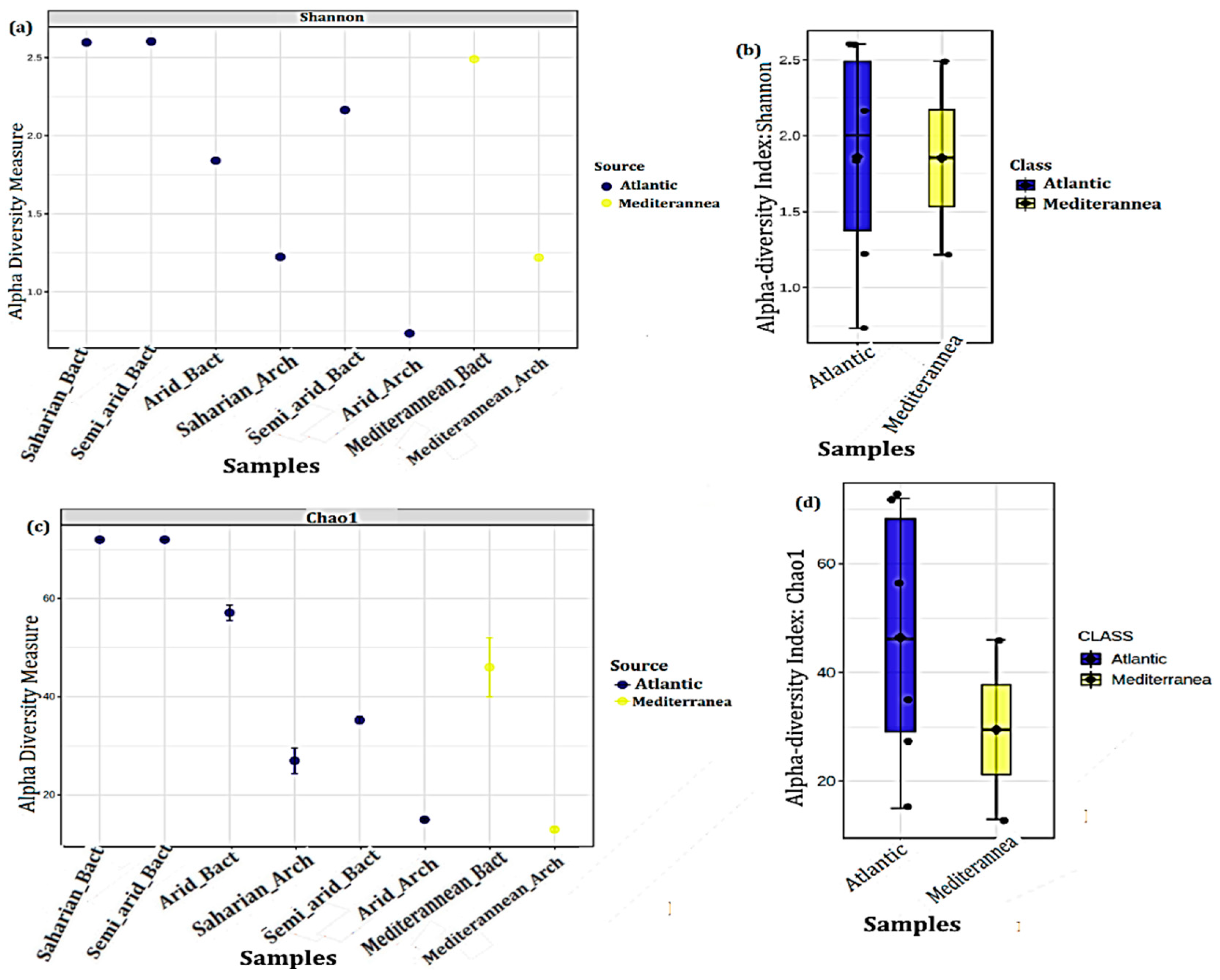
3.3.1. Alpha diversity
3.3.2. Beta Diversity
3.4. Hierarchically Clustered Heat Map
3.5. Core Microbiome Analysis
3.6. Correlation Analysis
3.7. Differential Abundance Analysis
3.8. Functional Profiling and Metabolic Network Visualization
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Falkowski, P.; Barber, R.; Smetacek, V. Biogeochemical Controls and Feedbacks on Ocean Primary Production. Science 1998, 281, 200–207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whitman, W.B.; Coleman, D.C.; Wiebe, W.J. Prokaryotes: The unseen majority. Proc. Natl. Acad. Sci. USA 1998, 95, 6578–6583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doney, S.C.; Ruckelshaus, M.; Emmett Duffy, J.; Barry, J.P.; Chan, F.; English, C.A.; Galindo, H.M.; Grebmeier, J.M.; Hollowed, A.B.; Knowlton, N.; et al. Climate Change Impacts on Marine Ecosystems. Annu. Rev. Mar. Sci. 2012, 4, 11–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steele, J.A.; Countway, P.D.; Xia, L.; Vigil, P.D.; Beman, J.M.; Kim, D.Y.; Chow, C.-E.T.; Sachdeva, R.; Jones, A.C.; Schwalbach, M.S.; et al. Marine bacterial, archaeal and protistan association networks reveal ecological linkages. ISME J. 2011, 5, 1414–1425. [Google Scholar] [CrossRef]
- Martiny, J.B.H.; Bohannan, B.J.M.; Brown, J.H.; Colwell, R.K.; Fuhrman, J.A.; Green, J.L.; Horner-Devine, M.C.; Kane, M.; Krumins, J.A.; Kuske, C.R.; et al. Microbial biogeography: Putting microorganisms on the map. Nat. Rev. Microbiol. 2006, 4, 102–112. [Google Scholar] [CrossRef] [PubMed]
- ISO-19458-2006.pdf. Consulté le: 13 Décembre 2021. [En Ligne]. Available online: https://cdn.standards.iteh.ai/samples/33845/be92aa0cd94d4a1b9148fce33d97dc59/ISO-19458-2006.pdf (accessed on 9 July 2022).
- Klindworth, A.; Pruesse, E.; Schweer, T.; Peplies, J.; Quast, C.; Horn, M.; Glöckner, F.O. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 2013, 41, e1. [Google Scholar] [CrossRef]
- Herlemann, D.P.; Labrenz, M.; Jürgens, K.; Bertilsson, S.; Waniek, J.J.; Andersson, A.F. Transitions in bacterial communities along the 2000 km salinity gradient of the Baltic Sea. ISME J. 2011, 5, 1571–1579. [Google Scholar] [CrossRef] [Green Version]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J.; et al. Introducing mothur: Open-Source, Platform-Independent, Community-Supported Software for Describing and Comparing Microbial Communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef] [Green Version]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef]
- Toumi, M.; Abbaszade, G.; Sbaoui, Y.; Farkas, R.; Acs, E.; Jurecska, L.; Tóth, E. Cultivation and Molecular Studies to Reveal the Microbial Communities of Groundwaters Discharge Located in Hungary. Water 2021, 13, 533. [Google Scholar] [CrossRef]
- Edgar, R.C.; Haas, B.J.; Clemente, J.C.; Quince, C.; Knight, R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 2011, 27, 2194–2200. [Google Scholar] [CrossRef] [Green Version]
- Ondov, B.D.; Bergman, N.H.; Phillippy, A.M. Interactive metagenomic visualization in a Web browser. BMC Bioinform. 2011, 12, 385. [Google Scholar] [CrossRef] [Green Version]
- Sbaoui, Y.; Nouadi, B.; Ezaouine, A.; Salam, R.; Elmessal, M.; Bennis, F.; Chegdani, F. Functional Prediction of Biological Profile During Eutrophication in Marine Environment. Bioinform. Biol. Insights 2022, 16, 117793222110639. [Google Scholar] [CrossRef] [PubMed]
- Friedman, J.; Alm, E.J. Inferring Correlation Networks from Genomic Survey Data. PLoS Comput. Biol. 2012, 8, e1002687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cavicchioli, R.; Ripple, W.J.; Timmis, K.N.; Azam, F.; Bakken, L.R.; Baylis, M.; Behrenfeld, M.J.; Boetius, A.; Boyd, P.W.; Classen, A.T.; et al. Scientists’ warning to humanity: Microorganisms and climate change. Nat. Rev. Microbiol. 2019, 17, 569–586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Behrenfeld, M.J. Climate-mediated dance of the plankton. Nat. Clim. Chang. 2014, 4, 880–887. [Google Scholar] [CrossRef]
- Magne, F.; Gotteland, M.; Gauthier, L.; Zazueta, A.; Pesoa, S.; Navarrete, P.; Balamurugan, R. The Firmicutes/Bacteroidetes Ratio: A Relevant Marker of Gut Dysbiosis in Obese Patients? Nutrients 2020, 12, 1474. [Google Scholar] [CrossRef] [PubMed]
- Siles, J.A.; Margesin, R. Abundance and Diversity of Bacterial, Archaeal, and Fungal Communities Along an Altitudinal Gradient in Alpine Forest Soils: What Are the Driving Factors? Microb. Ecol. 2016, 72, 207–220. [Google Scholar] [CrossRef] [Green Version]
- 1225 New York Avenue et NW. Algae, Cyanobacteria Blooms, and Climate Change. Climate Institute. Available online: http://climate.org/algae-cyanobacteria-blooms-and-climate-change/ (accessed on 6 December 2021).
- Oyama, Y.; Matsushita, B.; Fukushima, T. Cyanobacterial Blooms as an Indicator of Environmental Degradation in Waters and Their Monitoring Using Satellite Remote Sensing. In Aquatic Biodiversity Conservation and Ecosystem Services; Nakano, S., Yahara, T., Nakashizuka, T., Eds.; Ecological Research Monographs; Springer: Singapore, 2016; pp. 71–85. ISBN 978-981-10-0780-4. [Google Scholar]
- Sebastián, M.; Ortega-Retuerta, E.; Gómez-Consarnau, L.; Zamanillo, M.; Álvarez, M.; Arístegui, J.; Gasol, J.M. Environmental gradients and physical barriers drive the basin-wide spatial structuring of Mediterranean Sea and adjacent eastern Atlantic Ocean prokaryotic communities. Limnol. Oceanogr. 2021, 66, 4077–4095. [Google Scholar] [CrossRef]
- Giovannoni, S.J.; Stingl, U. Molecular diversity and ecology of microbial plankton. Nature 2005, 437, 343–348. [Google Scholar] [CrossRef]
- Vieira-Silva, S.; Rocha, E.P.C. The Systemic Imprint of Growth and Its Uses in Ecological (Meta) Genomics. PLoS Genet. 2010, 6, e1000808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sbaoui, Y.; Bennis, F.; Chegdani, F. SARS-CoV-2 as Enteric Virus in Wastewater: Which Risk on the Environment and Human Behavior? Microbiol. Insights 2021, 14, 1178636121999673. [Google Scholar] [CrossRef] [PubMed]
- Johnson, Z.I.; Zinser, E.R.; Coe, A.; McNulty, N.P.; Woodward, E.M.S.; Chisholm, S.W. Niche partitioning among Prochlorococcus ecotypes along ocean-scale environmental gradients. Science 2006, 311, 1737–1740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lozupone, C.A.; Knight, R. Global patterns in bacterial diversity. Proc. Natl. Acad. Sci. USA 2007, 104, 11436–11440. [Google Scholar] [CrossRef] [Green Version]
- Raes, J.; Letunic, I.; Yamada, T.; Jensen, L.J.; Bork, P. Toward molecular trait-based ecology through integration of biogeochemical, geographical and metagenomic data. Mol. Syst. Biol. 2011, 7, 473. [Google Scholar] [CrossRef] [PubMed]
- Habbane, M.; Montoya, J.; Rhouda, T.; Sbaoui, Y.; Radallah, D.; Emperador, S. Human Mitochondrial DNA: Particularities and Diseases. Biomedicines 2021, 9, 1364. [Google Scholar] [CrossRef] [PubMed]
- DeLong, E.F.; Preston, C.M.; Mincer, T.; Rich, V.; Hallam, S.J.; Frigaard, N.-U.; Martinez, A.; Sullivan, M.B.; Edwards, R.; Brito, B.R.; et al. Community genomics among stratified microbial assemblages in the ocean’s interior. Science 2006, 311, 496–503. [Google Scholar] [CrossRef]
- Martel, C.M.; Flynn, K.J. Morphological controls on cannibalism in a planktonic marine phagotroph. Protist 2008, 159, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Stocker, R. Marine Microbes See a Sea of Gradients. Science 2012, 338, 628–633. [Google Scholar] [CrossRef] [Green Version]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample ID | Saharian | Arid | Semi-Arid | Mediterranean |
|---|---|---|---|---|
| Carbon Monoxide (COSC); (µg/m3) | 138.69 | 140.87 | 144.86 | 147.04 |
| Carbon Dioxide (CO2sc); (mg/m3) | 774.59 | 775.62 | 780.11 | 787.34 |
| Sulfur Dioxide (SO2sm); (µg/m3) | 0.07 | 0.11 | 0.21 | 0.86 |
| Nitrogen Dioxide (NO2) | No data | No data | No data | No data |
| Dust Extinction (Aerosol Optical Thickness, 550 nm) (Duex) (T) | 0.1009 | 0.0044 | 0.0203 | 0.0278 |
| Particulate Matter < 1 µm (PM1); (µg/m3) | 1 | 1 | 1 | 1 |
| Particulate Matter < 2.5 µm (PM2.5); (µg/m3) | 6 | 3 | 4 | 4 |
| Particulate Matter < 10 µm (PM10); (µg/m3) | 21 | 8 | 9 | 21 |
| Sulfate Extinction (Aerosol Optical Thickness, 550 nm) (AOT) (SO4ex) (T) | 0.013 | 0.015 | 0.016 | 0.013 |
| Gene-Region | Phyla | Ref-Primer | Program | Reference | |
|---|---|---|---|---|---|
| 16S-V4 | Bacteria | F = CS1-TS-B341F | 98 °C for 3 min | [7] | |
| 25 Cycles | 95 °C for 30 s | ||||
| 55 °C for 30 s | |||||
| R = CS2-TS-805NR | 72 °C for 30 s | ||||
| 72 °C for 5 min | |||||
| 16S-V4 | Archaea | F = CS1-F-A519F | 98 °C for 3 min | [8] | |
| 25 Cycles | 95 °C for 30 s | ||||
| 60 °C for 30 s | |||||
| R = CS2-TS-Arch-855R | 72 °C for 30 s | ||||
| 72 °C for 10 min | |||||
| Sample ID | Saharian | Arid | Semi-Arid | Mediterranean |
|---|---|---|---|---|
| Temp; (°C) | 19.7 | 20 | 19 | 17 |
| pH | 8.15 | 8.15 | 8.3 | 8.3 |
| Salinity; (‰) | 37.5 | 37.1 | 37.6 | 41.2 |
| Dissolved oxygen; (mg/L) | 9.8 | 7.7 | 10.3 | 9.5 |
| Genus | r | p |
|---|---|---|
| Polaribacter | 0.651 | 0.0801 |
| Yoonia_Lokatanella | 0.647 | 0.0823 |
| Amylibacter | 0.635 | 0.0905 |
| Planktomarina | 0.623 | 0.0985 |
| SUP05_cluster | 0.6131 | 0.1059 |
| SAR86_clade_ge | 0.609 | 0.1089 |
| HIMB11 | 0.606 | 0.1110 |
| Aurantivigra | 0.594 | 0.1201 |
| NS4_marine_group | 0.561 | 0.1471 |
| Synechococcus_CC9902 | 0.2646 | 0.0297 |
| Marinimicrobia_ge | −0.27 | 0.0396 |
| Roseburia | 0.536 | 0.170 |
| Pseudoalteromonadaceae | 0.436 | 0.2796 |
| Clade_la | 0.408 | 0.3147 |
| Pseudoalteromonas | −0.430 | 0.2864 |
| Akkermansia | −0.437 | 0.2787 |
| Butyrivibrio | −0.439 | 0.276 |
| RC9_gut_group | −0.482 | 0.2257 |
| Phascolarctobacter | −0.495 | 0.2118 |
| Alistipes | −0.5116 | 0.1949 |
| Solibacillus | −0.560 | 0.148 |
| Bacteroides | −0.572 | 0.1376 |
| Coprococcus | −0.577 | 0.1336 |
| Incertae_Sedis | −0.602 | 0.114 |
| Uncultured_bacteria | −0.630 | 0.0940 |
| log2FC | logCPM | p-Values | FDR | |
|---|---|---|---|---|
| Polaribacter | 7.6426 | 14.113 | 3.0624 × 10−5 | 0.0040577 |
| Yoonia_Loktanella | 6.9335 | 14.034 | 5.1361 × 10−5 | 0.0045369 |
| Amylibacter | 6.8861 | 15.819 | 0.00029882 | 0.012863 |
| Rhodobacteraceae_unclassified | 5.8166 | 12.39 | 0.00033977 | 0.012863 |
| SAR86_clade_ge | 5.625 | 13.232 | 0.00050154 | 0.016613 |
| Rhodobacteraceae_unclassified_5 | 4.2462 | 10.795 | 0.00065675 | 0.018107 |
| Aurantivirga | 5.7707 | 14.009 | 0.00068327 | 0.018107 |
| Planktomarina | 4.9286 | 11.689 | 0.00081746 | 0.019693 |
| HIMB11 | 5.2131 | 12.78 | 0.00091929 | 0.020301 |
| SUP05_cluster | 4.6165 | 11.331 | 0.0012203 | 0.023696 |
| Candidatus_Actinomarina_1 | 3.9349 | 10.722 | 0.0012519 | 0.023696 |
| Synechococcus_CC9902 | 3.8306 | 11.186 | 0.0019083 | 0.031766 |
| NS4_marine_group | 4.1886 | 11.221 | 0.0019179 | 0.031766 |
| SAR86_clade_ge_1 | 4.4104 | 11.766 | 0.0025391 | 0.03958 |
| Marinimicrobia__ge | 4.1083 | 11.494 | 0.0034288 | 0.047822 |
| Enterobacterales_unclassified_3 | 3.0121 | 10.025 | 0.0048204 | 0.062794 |
| Pseudoalteromonadaceae_unclassified_5 | 2.9056 | 10.043 | 0.0049761 | 0.062794 |
| Acinetobacter | 2.6463 | 10.089 | 0.007196 | 0.082911 |
| Clade_I_unclassified | 2.5445 | 10.117 | 0.010326 | 0.11401 |
| Marine_Group_II_ge_6 | 4.0622 | 11.844 | 0.011044 | 0.11539 |
| Rhodobacteraceae_unclassified_4 | 3.0368 | 11.103 | 0.011321 | 0.11539 |
| Marine_Group_II_ge_10 | 3.0639 | 10.456 | 0.013922 | 0.13664 |
| Sphingorhabdus | 2.4657 | 9.9038 | 0.015954 | 0.151 |
| Rhodobacteraceae_unclassified_1 | 3.6972 | 12.13 | 0.017132 | 0.15656 |
| Clade_Ia | 4.1302 | 15.278 | 0.019241 | 0.16996 |
| Marine_Group_II_ge_9 | 2.6756 | 10.447 | 0.029006 | 0.24795 |
| Candidatus_Puniceispirillum | 2.4428 | 10.843 | 0.029999 | 0.24843 |
| NS5_marine_group | 3.2383 | 12.426 | 0.032082 | 0.25763 |
| OM60_clade_1 | 2.3647 | 10.552 | 0.036781 | 0.28667 |
| Woesearchaeia_ge_1 | 2.5099 | 10.189 | 0.04154 | 0.31452 |
| Glaciecola | 3.2737 | 12.479 | 0.046068 | 0.3365 |
| Clade_II_ge | 2.7034 | 11.728 | 0.048253 | 0.3365 |
| Marine_Group_II_ge_8 | 2.4898 | 10.685 | 0.053948 | 0.35414 |
| Pseudohongiella | 2.0693 | 9.8599 | 0.054751 | 0.35414 |
| Marine_Group_II_ge_7 | 3.0471 | 11.682 | 0.063485 | 0.40056 |
| Candidatus_Actinomarina | 2.5996 | 11.999 | 0.071843 | 0.44276 |
| Candidatus_Nitrosopumilus_3 | 2.332 | 10.727 | 0.073943 | 0.44534 |
| Woesearchaeia_ge | 2.2609 | 10.747 | 0.09075 | 0.5228 |
| Marine_Group_II_ge_3 | 2.7335 | 12.328 | 0.099505 | 0.56104 |
| Marine_Group_II_ge_4 | 2.6089 | 12.202 | 0.1147 | 0.63322 |
| Pseudoalteromonas_1 | −5.2091 | 15.565 | 0.13898 | 0.75162 |
| Marine_Group_II_ge_11 | 1.9035 | 10.397 | 0.15393 | 0.81582 |
| Thermoplasmata_unclassified | 1.715 | 10.45 | 0.18448 | 0.95639 |
| Marine_Group_II_ge_2 | 2.2069 | 12.934 | 0.18767 | 0.95639 |
| Marine_Group_II_ge_5 | 2.0212 | 12.197 | 0.20278 | 0.98455 |
| Flavobacteriaceae_unclassified | 1.1644 | 9.7761 | 0.22177 | 1 |
| Pseudoalteromonas | −4.3703 | 16.223 | 0.23593 | 1 |
| Marine_Group_II_ge_1 | 2.032 | 15.231 | 0.245 | 1 |
| Marine_Group_II_ge_14 | 1.4912 | 10.295 | 0.26058 | 1 |
| NS5_marine_group_1 | 1.062 | 10.659 | 0.28825 | 1 |
| Candidatus_Nitrosopelagicus | 1.4629 | 13.514 | 0.3687 | 1 |
| Cobetia | −2.7752 | 13.03 | 0.37506 | 1 |
| Nitrosopumilaceae_unclassified | 1.3204 | 13.01 | 0.39378 | 1 |
| Candidatus_Nitrosopumilus_2 | 1.2182 | 10.994 | 0.39482 | 1 |
| Pseudoalteromonas_2 | −1.9949 | 10.992 | 0.42023 | 1 |
| Rhodobacteraceae_unclassified_3 | 0.92903 | 10.463 | 0.42875 | 1 |
| Psychrobacter | −2.6193 | 12.888 | 0.45942 | 1 |
| Pseudoalteromonas_10 | 0.68628 | 9.712 | 0.47556 | 1 |
| Nitrosopumilaceae_unclassified_2 | 0.88441 | 10.555 | 0.4964 | 1 |
| Vibrionaceae_unclassified | −1.8098 | 11.329 | 0.51009 | 1 |
| Marine_Group_II_ge | 0.97786 | 16.549 | 0.51011 | 1 |
| Shewanella | −1.5 | 10.617 | 0.55035 | 1 |
| Nitrosopumilaceae_unclassified_4 | 0.78774 | 10.43 | 0.55367 | 1 |
| Nitrosopumilaceae_unclassified_1 | 0.64499 | 12.682 | 0.59802 | 1 |
| Bacteria_unclassified_5 | −1.2119 | 10.405 | 0.61273 | 1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sbaoui, Y.; Ezaouine, A.; Toumi, M.; Farkas, R.; Kbaich, M.A.; Habbane, M.; El Mouttaqui, S.; Kadiri, F.Z.; El Messal, M.; Tóth, E.; et al. Effect of Climate on Bacterial and Archaeal Diversity of Moroccan Marine Microbiota. Microorganisms 2022, 10, 1622. https://doi.org/10.3390/microorganisms10081622
Sbaoui Y, Ezaouine A, Toumi M, Farkas R, Kbaich MA, Habbane M, El Mouttaqui S, Kadiri FZ, El Messal M, Tóth E, et al. Effect of Climate on Bacterial and Archaeal Diversity of Moroccan Marine Microbiota. Microorganisms. 2022; 10(8):1622. https://doi.org/10.3390/microorganisms10081622
Chicago/Turabian StyleSbaoui, Yousra, Abdelkarim Ezaouine, Marwene Toumi, Rózsa Farkas, Mouad Ait Kbaich, Mouna Habbane, Sara El Mouttaqui, Fatem Zahra Kadiri, Mariame El Messal, Erika Tóth, and et al. 2022. "Effect of Climate on Bacterial and Archaeal Diversity of Moroccan Marine Microbiota" Microorganisms 10, no. 8: 1622. https://doi.org/10.3390/microorganisms10081622