
The Effects of DNA Extraction Kits and Primers on Prokaryotic and Eukaryotic Microbial Community in Freshwater Sediments

 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection and Processing
2.2. DNA Extraction, PCR and Sequencing
2.3. Sequence Data Processing and Taxonomic Assignment
2.4. Statistical Analysis
3. Results
3.1. Effects of DNA Extraction Kits in DNA Yield
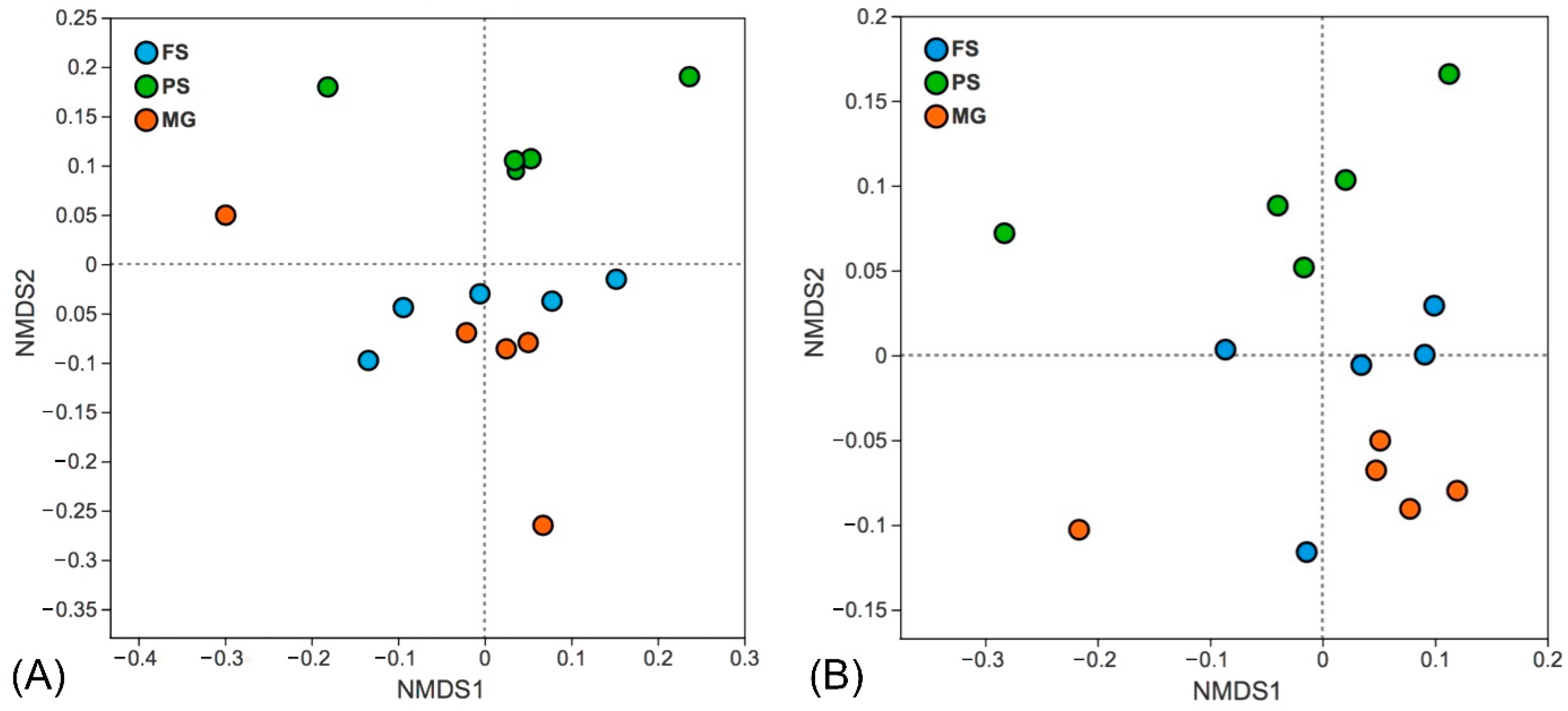
3.2. Effects of DNA Extraction Kits on Prokaryotic Microbial Community
3.3. Effects of DNA Extraction Kits and Primer Sets on Eukaryotic Microbial Community
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shokralla, S.; Spall, J.L.; Gibson, J.F.; Hajibabaei, M. Next-generation sequencing technologies for environmental DNA research. Mol. Ecol. 2012, 21, 1794–1805. [Google Scholar] [CrossRef] [PubMed]
- Geisen, S.; Laros, I.; Vizcaíno, A.; Bonkowski, M.; De Groot, G. Not all are free-living: High-throughput DNA metabarcoding reveals a diverse community of protists parasitizing soil metazoa. Mol. Ecol. 2015, 24, 4556–4569. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.X.; Liu, F.H.; Zheng, S.L.; Chen, L.; Zhang, X.L.; Gong, J. The differentiation of iron-reducing bacterial community and iron-reduction activity between riverine and marine sediments in the Yellow River estuary. Mar. Life Sci. Technol. 2020, 2, 87–96. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.F.; Xiao, X.; Zhang, Y. Microbial diversity of sediments from an inactive hydrothermal vent field, southwest Indian ridge. Mar. Life Sci. Technol. 2020, 2, 73–86. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Zhao, Q.N.; McMinn, A.; Yang, E.J.; Jiang, Y. Planktonic microbial eukaryotes in polar surface waters: Recent advances in high-throughput sequencing. Mar. Life Sci. Technol. 2020, 3, 94–102. [Google Scholar] [CrossRef]
- Capo, E.; Ninnes, S.; Domaizon, I.; Bertilsson, S.; Bigler, C.; Wang, X.; Bindler, R.; Rydberg, J. Landscape setting drives the microbial eukaryotic community structure in four swedish mountain lakes over the Holocene. Microorganisms 2021, 9, 355. [Google Scholar] [CrossRef]
- Guo, F.; Zhang, T. Biases during DNA extraction of activated sludge samples revealed by high throughput sequencing. Appl. Microbiol. Biotechnol. 2013, 97, 4607–4616. [Google Scholar] [CrossRef] [Green Version]
- Mirsepasi, H.; Persson, S.; Struve, C.; Andersen, L.O.; Petersen, A.M.; Krogfelt, K.A. Microbial diversity in fecal samples depends on DNA extraction method: easyMag DNA extraction compared to QIAamp DNA stool mini kit extraction. BMC Res. Notes 2014, 7, 50. [Google Scholar] [CrossRef] [Green Version]
- Gohl, D.M.; Vangay, P.; Garbe, J.; MacLean, A.; Hauge, A.; Becker, A.; Gould, T.J.; Clayton, J.B.; Johnson, T.J.; Hunter, R.; et al. Systematic improvement of amplicon marker gene methods for increased accuracy in microbiome studies. Nat. Biotechnol. 2016, 34, 942–949. [Google Scholar] [CrossRef] [Green Version]
- Costea, P.I.; Zeller, G.; Sunagawa, S.; Pelletier, E.; Alberti, A.; Levenez, F.; Tramontano, M.; Driessen, M.; Hercog, R.; Jung, F.E. Towards standards for human fecal sample processing in metagenomic studies. Nat. Biotechnol. 2017, 35, 1069–1076. [Google Scholar] [CrossRef]
- Sinha, R.; Abu-Ali, G.; Vogtmann, E.; Fodor, A.A.; Ren, B.; Amir, A.; Schwager, E.; Crabtree, J.; Ma, S.; Microbiome Quality Control Project Consortium; et al. Assessment of variation in microbial community amplicon sequencing by the Microbiome Quality Control (MBQC) project consortium. Nat. Biotechnol. 2017, 35, 1077–1086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quince, C.; Walker, A.W.; Simpson, J.T.; Loman, N.J.; Segata, N. Shotgun metagenomics, from sampling to analysis. Nat. Biotechnol. 2017, 35, 833–844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, S.; Bunge, J.; Leslin, C.; Jeon, S.; Epstein, S.S. Polymerase chain reaction primers miss half of rRNA microbial diversity. ISME J. 2009, 3, 1365–1373. [Google Scholar] [CrossRef] [PubMed]
- Stach, J.E.; Bathe, S.; Clapp, J.P.; Burns, R.G. PCR-SSCP comparison of 16S rDNA sequence diversity in soil DNA obtained using different isolation and purification methods. FEMS Microbiol. Ecol. 2001, 36, 139–151. [Google Scholar] [CrossRef] [PubMed]
- Martincová, I.; Aghová, T. Comparison of 12 DNA extraction kits for vertebrate samples. Anim. Biodiv. Conserv. 2020, 43, 67. [Google Scholar] [CrossRef] [Green Version]
- Burbach, K.; Seifert, J.; Pieper, D.H.; Camarinha-Silva, A. Evaluation of DNA extraction kits and phylogenetic diversity of the porcine gastrointestinal tract based on Illumina sequencing of two hypervariable regions. Microbiologyopen 2016, 5, 70–82. [Google Scholar] [CrossRef]
- Ramírez, G.A.; Graham, D.; D’Hondt, S. Influence of commercial DNA extraction kit choice on prokaryotic community metrics in marine sediment. Limmol. Oceanogr. Methods 2018, 16, 525–536. [Google Scholar] [CrossRef]
- Wesolowska-Andersen, A.; Bahl, M.I.; Carvalho, V.; Kristiansen, K.; Sicheritz-Pontén, T.; Gupta, R.; Licht, T.R. Choice of bacterial DNA extraction method from fecal material influences community structure as evaluated by metagenomic analysis. Microbiome 2014, 2, 19. [Google Scholar] [CrossRef] [Green Version]
- Teng, F.; Darveekaran Nair, S.S.; Zhu, P.; Li, S.; Huang, S.; Li, X.; Xu, J.; Yang, F. Impact of DNA extraction method and targeted 16S-rRNA hypervariable region on oral microbiota profiling. Sci. Rep. 2018, 8, 16321. [Google Scholar] [CrossRef]
- Mackenzie, B.W.; Waite, D.W.; Taylor, M.W. Evaluating variation in human gut microbiota profiles due to DNA extraction method and inter-subject difference. Front. Microbiol. 2015, 6, 130. [Google Scholar] [CrossRef] [Green Version]
- Desneux, J.; Pourcher, A.M. Comparison of DNA extraction kits and modification of DNA elution procedure for the quantitation of subdominant bacteria from piggery effluents with real-time PCR. Microbiologyopen 2014, 3, 437–445. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Xue, Y.; Yang, J. Rare plankton subcommunities are far more affected by DNA extraction kits than abundant plankton. Front. Microbiol. 2019, 10, 454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mao, D.P.; Zhou, Q.; Chen, C.Y.; Quan, Z.X. Coverage evaluation of universal bacterial primers using the metagenomic datasets. BMC Microbiol. 2012, 12, 66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hadziavdic, K.; Lekang, K.; Lanzen, A.; Jonassen, I.; Thompson, E.M.; Troedsson, C. Characterization of the 18S rRNA gene for designing universal eukaryote specific primers. PLoS ONE 2014, 9, e87624. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.N.; Feng, K.; Li, S.Z.; Wang, Z.J.; Zhang, Z.J.; Deng, Y. In-silico evaluation and improvement on 16S/18S/ITS primers for amplicon high-throughput sequencing. Microbiology 2020, 47, 2897–2912. [Google Scholar] [CrossRef]
- Apprill, A.; McNally, S.; Parsons, R.; Weber, L. Minor revision to V4 region SSU rRNA 806R gene primer greatly increases detection of SAR11 bacterioplankton. Aquat. Microb. Ecol. 2015, 75, 129–137. [Google Scholar] [CrossRef] [Green Version]
- Caporaso, J.G.; Lauber, C.L.; Walters, W.A.; Berg-Lyons, D.; Lozupone, C.A.; Turnbaugh, P.J.; Fierer, N.; Knight, R. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc. Natl. Acad. Sci. USA 2011, 108 (Suppl. S1), 4516–4522. [Google Scholar] [CrossRef] [Green Version]
- Stoeck, T.; Bass, D.; Nebel, M.; Christen, R.; Jones, M.D.; Breiner, H.W.; Richards, T.A. Multiple marker parallel tag environmental DNA sequencing reveals a highly complex eukaryotic community in marine anoxic water. Mol. Ecol. 2010, 19 (Suppl. S1), 21–31. [Google Scholar] [CrossRef]
- Simon, M.; Jardillier, L.; Deschamps, P.; Moreira, D.; Restoux, G.; Bertolino, P.; Lopez-Garcia, P. Complex communities of small protists and unexpected occurrence of typical marine lineages in shallow freshwater systems. Environ. Microbiol. 2015, 17, 3610–3627. [Google Scholar] [CrossRef] [Green Version]
- Li, X.H.; Huang, J.; Filker, S.; Stoeck, T.; Bi, Y.H.; Yu, Y.H.; Song, W.B. Spatio-temporal patterns of zooplankton in a main-stem dam affected tributary: A case study in the Xiangxi River of the Three Gorges Reservoir, China. Sci. China Life Sci. 2019, 62, 1058–1069. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magoč, T.; Salzberg, S.L. FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C.; Haas, B.J.; Clemente, J.C.; Quince, C.; Knight, R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 2011, 27, 2194–2200. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.C. UPARSE: Highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 2013, 10, 996–998. [Google Scholar] [CrossRef]
- Albertsen, M.; Karst, S.M.; Ziegler, A.S.; Kirkegaard, R.H.; Nielsen, P.H. Back to basics-the influence of DNA extraction and primer choice on phylogenetic analysis of activated sludge communities. PLoS ONE 2015, 10, e0132783. [Google Scholar] [CrossRef]
- Pollock, J.; Glendinning, L.; Wisedchanwet, T.; Watson, M. The madness of microbiome: Attempting to find consensus “best practice” for 16S microbiome studies. Appl. Environ. Microbiol. 2018, 84, e02627-17. [Google Scholar] [CrossRef] [Green Version]
- Kennedy, N.A.; Walker, A.W.; Berry, S.H.; Duncan, S.H.; Farquarson, F.M.; Louis, P.; Thomson, J.M.; UK IBD Genetics Consortium; Satsangi, J.; Flint, H.J.; et al. The impact of different DNA extraction kits and laboratories upon the assessment of human gut microbiota composition by 16S rRNA gene sequencing. PLoS ONE 2014, 9, e88982. [Google Scholar] [CrossRef]
- Gerasimidis, K.; Bertz, M.; Quince, C.; Brunner, K.; Bruce, A.; Combet, E.; Calus, S.; Loman, N.; Ijaz, U.Z. The effect of DNA extraction methodology on gut microbiota research applications. BMC Res. Notes 2016, 9, 365. [Google Scholar] [CrossRef] [Green Version]
- Henderson, G.; Cox, F.; Kittelmann, S.; Miri, V.H.; Zethof, M.; Noel, S.J.; Waghorn, G.C.; Janssen, P.H. Effect of DNA extraction methods and sampling techniques on the apparent structure of cow and sheep rumen microbial communities. PLoS ONE 2013, 8, e74787. [Google Scholar] [CrossRef] [Green Version]
- Brandt, J.; Albertsen, M. Investigation of detection limits and the influence of DNA extraction and primer choice on the observed microbial communities in drinking water samples using 16S rRNA gene amplicon sequencing. Front. Microbiol. 2018, 9, 2140. [Google Scholar] [CrossRef]
- Carrigg, C.; Rice, O.; Kavanagh, S.; Collins, G.; O’Flaherty, V. DNA extraction method affects microbial community profiles from soils and sediment. Appl. Microbiol. Biotechnol. 2007, 77, 955–964. [Google Scholar] [CrossRef] [PubMed]
- Vesty, A.; Biswas, K.; Taylor, M.W.; Gear, K.; Douglas, R.G. Evaluating the impact of DNA extraction method on the representation of human oral bacterial and fungal communities. PLoS ONE 2017, 12, e0169877. [Google Scholar] [CrossRef] [PubMed]
- Dineen, S.M.; Aranda, R.; Anders, D.L.; Robertson, J.M. An evaluation of commercial DNA extraction kits for the isolation of bacterial spore DNA from soil. J. Appl. Microbiol. 2010, 109, 1886–1896. [Google Scholar] [CrossRef]
- Vishnivetskaya, T.A.; Layton, A.C.; Lau, M.C.; Chauhan, A.; Cheng, K.R.; Meyers, A.J.; Murphy, J.R.; Rogers, A.W.; Saarunya, G.S.; Williams, D.E.; et al. Commercial DNA extraction kits impact observed microbial community composition in permafrost samples. FEMS Microbiol. Ecol. 2014, 87, 217–230. [Google Scholar] [CrossRef] [PubMed]
- Multinu, F.; Harrington, S.C.; Chen, J.; Jeraldo, P.R.; Johnson, S.; Chia, N.; Walther-Antonio, M.R. Systematic bias introduced by genomic DNA template dilution in 16S rRNA gene-targeted microbiota profiling in human stool homogenates. mSphere 2018, 3, e00560-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polz, M.F.; Cavanaugh, C.M. Bias in template-toproduct ratios in multitemplate PCR. Appl. Environ. Microbiol. 1998, 64, 3724–3730. [Google Scholar] [CrossRef] [Green Version]
- Sipos, R.; Szekely, A.J.; Palatinszky, M.; Revesz, S.; Marialigeti, K.; Nikolausz, M. Effect of primer mismatch, annealing temperature and PCR cycle number on 16S rRNA gene-targeting bacterial community analysis. FEMS Microbiol. Ecol. 2007, 60, 341–350. [Google Scholar] [CrossRef] [Green Version]
- Kleter, B.; van Doorn, L.J.; ter Schegget, J.; Schrauwen, L.; van Krimpen, K.; Burger, M.; ter Harmsel, B.; Quint, W. Novel short-fragment PCR assay for highly sensitive broad-spectrum detection of anogenital human papillomaviruses. Am. J. Pathol. 1998, 153, 1731–1739. [Google Scholar] [CrossRef] [Green Version]
- Becker, S.; Boger, P.; Oehlmann, R.; Ernst, A. PCR bias in ecological analysis: A case study for quantitative Taq nuclease assays in analyses of microbial communities. Appl. Environ. Microbiol. 2000, 66, 4945–4953. [Google Scholar] [CrossRef] [Green Version]
- Huber, J.A.; Morrison, H.G.; Huse, S.M.; Neal, P.R.; Sogin, M.L.; Welch, D.B.M. Effect of PCR amplicon size on assessments of clone library microbial diversity and community structure. Environ. Microbiol. 2009, 11, 1292–1302. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Extraction Kit (Replicate) | DNA Yield (ng/μL) | A260/A280 | A260/A230 | 16S | 18S (Primer EK) | 18S (Primer TAR) | |||
|---|---|---|---|---|---|---|---|---|---|
| Observed OTUs | Number of Reads | Observed OTUs | Number of Reads | Observed OTUs | Number of Reads | ||||
| PS Kit (1) | 12.1 | 2.12 | 2.28 | 3447 | 53,722 | 464 | 54,167 | 495 | 56,794 |
| PS Kit (2) | 10.7 | 2.08 | 0.91 | 2918 | 47,465 | 373 | 31,698 | 452 | 47,406 |
| PS Kit (3) | 14.8 | 1.77 | 0.85 | 3569 | 58,878 | 414 | 26,946 | 723 | 32,269 |
| PS Kit (4) | 9.70 | 2.44 | 1.96 | 3063 | 37,905 | 389 | 34,004 | 446 | 68,706 |
| PS Kit (5) | 11.00 | 2.06 | 1.93 | 3442 | 64,948 | 426 | 41,928 | 534 | 56,171 |
| FS Kit (1) | 15.00 | 1.72 | 0.03 | 3082 | 40,066 | 332 | 25,834 | 659 | 59,622 |
| FS Kit (2) | 14.40 | 1.95 | 0.04 | 3244 | 33,726 | 291 | 26,791 | 975 | 63,743 |
| FS Kit (8) | 15.00 | 1.83 | 0.06 | 3349 | 38,358 | 307 | 42,675 | 355 | 66,368 |
| FS Kit (4) | 17.00 | 1.85 | 0.04 | 3785 | 62,632 | 342 | 42,608 | 904 | 54,906 |
| FS Kit (5) | 25.30 | 1.53 | 0.75 | 3095 | 36,319 | 324 | 33,241 | 406 | 30,630 |
| MG Kit (1) | 1.80 | 2.67 | 1.99 | 3254 | 51,613 | 168 | 43,146 | 146 | 50,094 |
| MG Kit (2) | 3.00 | 2.41 | 2.55 | 4072 | 64,839 | 160 | 58,909 | 92 | 21,079 |
| MG Kit (3) | 1.10 | 2.08 | 0.02 | 4129 | 67,204 | 156 | 62,365 | 86 | 32,158 |
| MG Kit (4) | 1.20 | 2.06 | 1.78 | 3541 | 41,225 | 109 | 28,986 | 67 | 56,659 |
| MG Kit (5) | 0.70 | 1.96 | 1.98 | 3950 | 52,999 | 115 | 27,190 | 108 | 48,887 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shi, Z.; Kong, Q.; Li, X.; Xu, W.; Mao, C.; Wang, Y.; Song, W.; Huang, J. The Effects of DNA Extraction Kits and Primers on Prokaryotic and Eukaryotic Microbial Community in Freshwater Sediments. Microorganisms 2022, 10, 1213. https://doi.org/10.3390/microorganisms10061213
Shi Z, Kong Q, Li X, Xu W, Mao C, Wang Y, Song W, Huang J. The Effects of DNA Extraction Kits and Primers on Prokaryotic and Eukaryotic Microbial Community in Freshwater Sediments. Microorganisms. 2022; 10(6):1213. https://doi.org/10.3390/microorganisms10061213
Chicago/Turabian StyleShi, Zihan, Qiaoyi Kong, Xinghao Li, Wenxin Xu, Chengzhi Mao, Yunfeng Wang, Weibo Song, and Jie Huang. 2022. "The Effects of DNA Extraction Kits and Primers on Prokaryotic and Eukaryotic Microbial Community in Freshwater Sediments" Microorganisms 10, no. 6: 1213. https://doi.org/10.3390/microorganisms10061213