
Comparative Analysis of Original and Replaced Gut Microbiomes within Same Individuals Identified the Intestinal Microbes Associated with Weight Gaining

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Design and Animal Experiments
2.2. Biochemical Analysis
2.3. Micro-Computed Tomography (Micro-CT)
2.4. Histological Analysis
2.5. 16S rRNA Gene Sequencing
2.6. Data Analyses
2.7. The α-Diversity Analysis for Relative Abundance Evaluation of Material and Microbiome
2.8. The β-Analysis for Relative Abundance Evaluation of Material and Microbiome
2.9. Establishment of Heatmap and Phylogenetic Tree
2.10. Co-Occurrence Network Construction
2.11. Statistical Analysis and Quantification
2.12. Ethics Approval
3. Results
3.1. Random Replacement of the Gut Microbiome of Conventional Mice with the Subset of the Human Gut Microbiome Affected Body Weight Gain Pattern
3.2. Different Kinds of Gut Microbiome Were Replaced in Each of the Experimental Mice
3.3. The Differences in Body Weight Gain Were Associated with the Different Shifts in the Composition of the Gut Microbiome
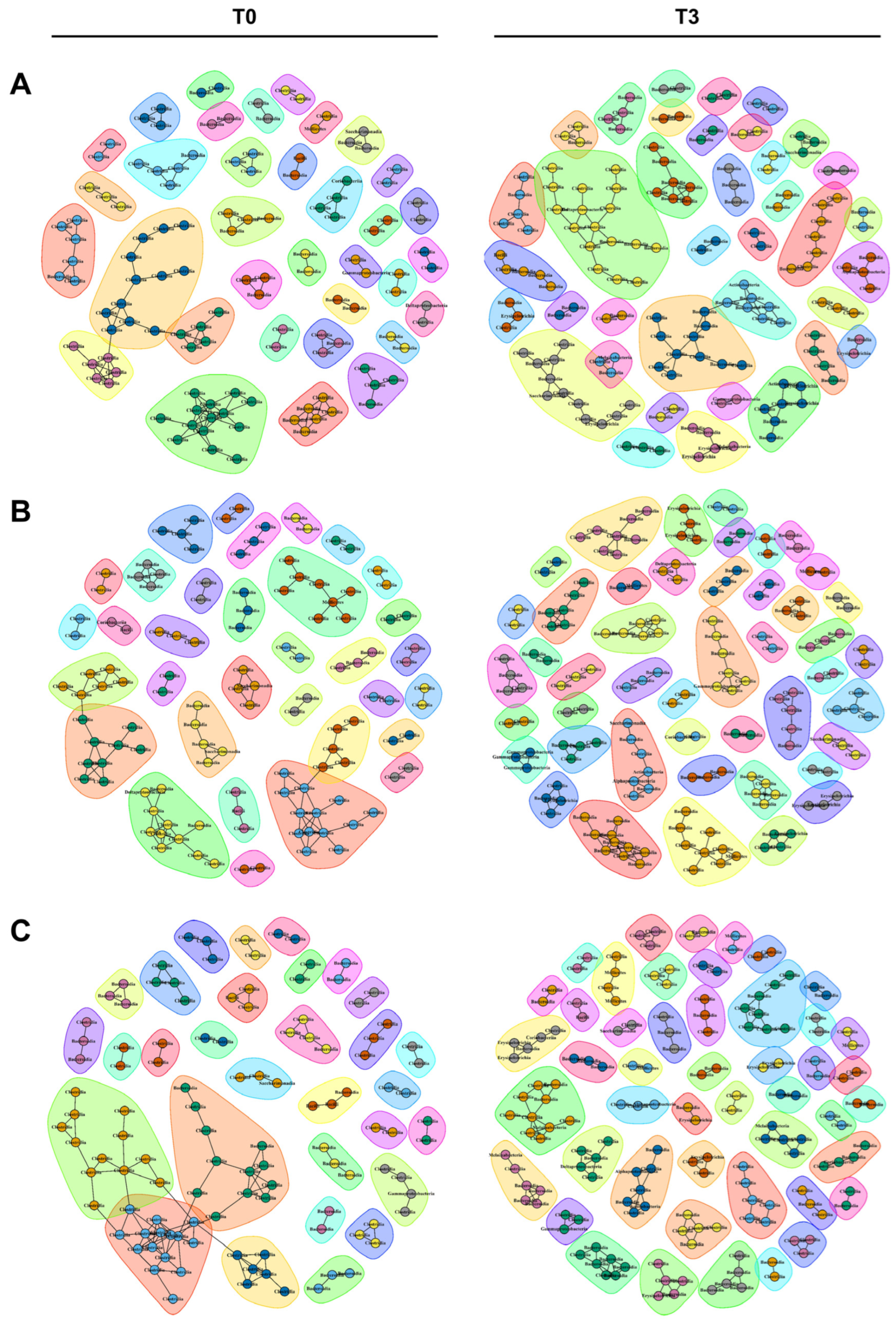
3.4. Different Kinds of Microbial Communities Were Established among the Three Groups of Mice
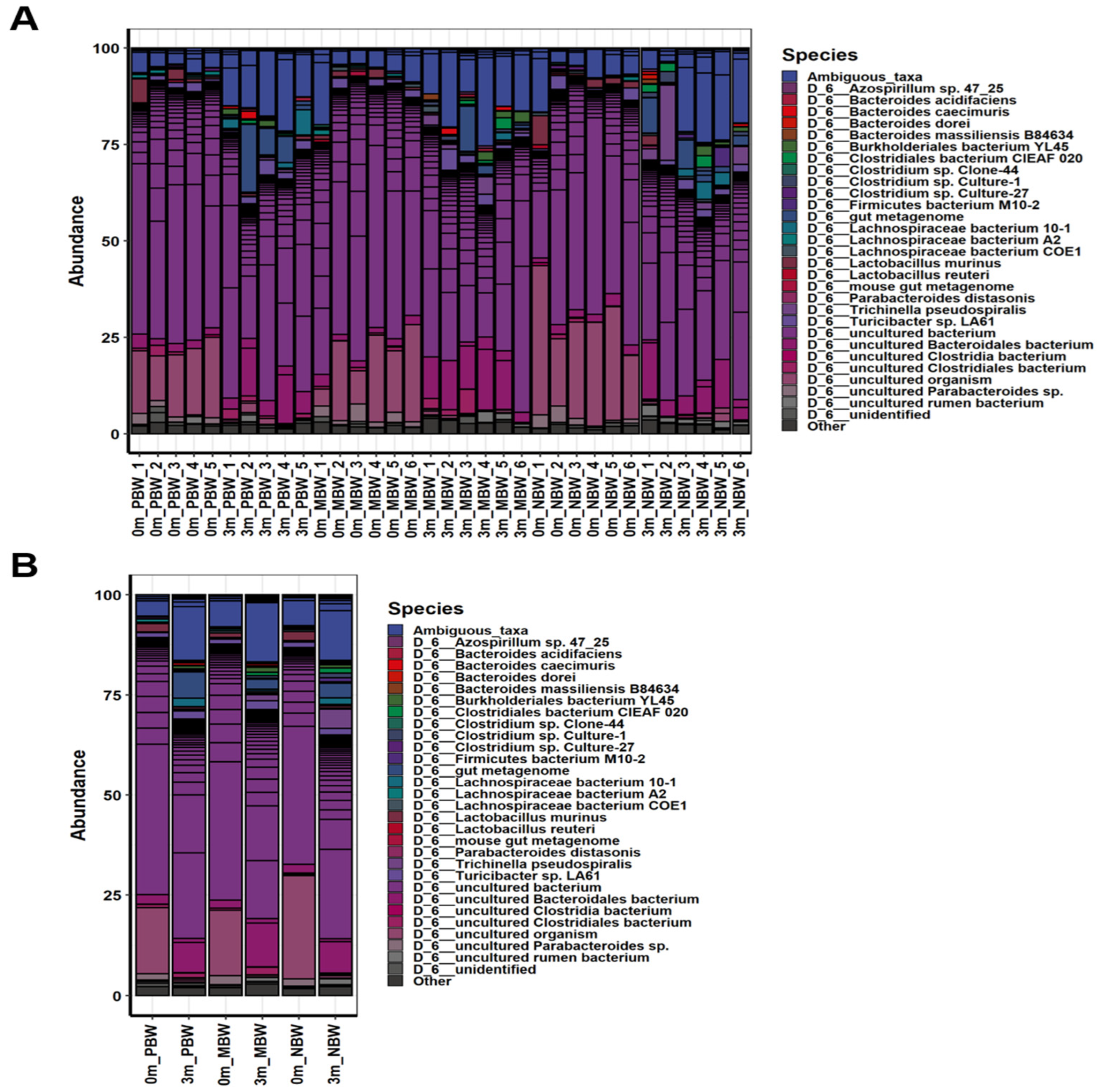
3.5. Intestinal Microbes That Affect Body Weight Gain Were Identified at the Species Level
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chen, X.; D’Souza, R.; Hong, S.T. The role of gut microbiota in the gut-brain axis: Current challenges and perspectives. Protein Cell 2013, 4, 403–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Lkhagva, E.; Chung, H.J.; Kim, H.J.; Hong, S.T. The pharmabiotic approach to treat hyperammonemia. Nutrients 2018, 10, 140. [Google Scholar] [CrossRef] [Green Version]
- Chung, H.J.; Nguyen, T.T.B.; Kim, H.J.; Hong, S.T. Gut Microbiota as a Missing Link Between Nutrients and Traits of Human. Front. Microbiol. 2018, 9, 1510. [Google Scholar] [CrossRef] [Green Version]
- Lkhagva, E.; Chung, H.J.; Ahn, J.S.; Hong, S.T. Host factors affect the gut microbiome more significantly than diet shift. Microorganisms 2021, 9, 2520. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.T.B.; Chung, H.J.; Kim, H.J.; Hong, S.T. Establishment of an ideal gut microbiota to boost healthy growth of neonates. Crit. Rev. Microbiol. 2019, 45, 118–129. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.T.B.; Jin, Y.Y.; Chung, H.J.; Hong, S.T. Pharmabiotics as an Emerging Medication for Metabolic Syndrome and Its Related Diseases. Molecules 2017, 22, 1795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ley, R.E.; Bäckhed, F.; Turnbaugh, P.; Lozupone, C.A.; Knight, R.D.; Gordon, J.I. Obesity alters gut microbial ecology. Proc. Natl. Acad. Sci. USA 2005, 102, 11070–11075. [Google Scholar] [CrossRef] [Green Version]
- Turnbaugh, P.J.; Hamady, M.; Yatsunenko, T.; Cantarel, B.L.; Duncan, A.; Ley, R.E.; Sogin, M.L.; Jones, W.J.; Roe, B.A.; Affourtit, J.P.; et al. A core gut microbiome in obese and lean twins. Nature 2009, 457, 480–484. [Google Scholar] [CrossRef] [Green Version]
- Xu, P.; Li, M.; Zhang, J.; Zhang, T. Correlation of intestinal microbiota with overweight and obesity in Kazakh school children. BMC Microbiol. 2012, 12, 283. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; DiBaise, J.K.; Zuccolo, A.; Kudrna, D.; Braidotti, M.; Yu, Y.; Parameswaran, P.; Crowell, M.D.; Wing, R.; Rittmann, B.E.; et al. Human gut microbiota in obesity and after gastric bypass. Proc. Natl. Acad. Sci. USA 2009, 106, 2365–2370. [Google Scholar] [CrossRef] [Green Version]
- Duncan, S.H.; Lobley, G.E.; Holtrop, G.; Ince, J.; Johnstone, A.M.; Louis, P.; Flint, H.J. Human colonic microbiota associated with diet, obesity and weight loss. Int. J. Obes. 2008, 32, 1720–1724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwiertz, A.; Taras, D.; Schäfer, K.; Beijer, S.; Bos, N.A.; Donus, C.; Hardt, P.D. Microbiota and SCFA in lean and overweight healthy subjects. Obesity 2010, 18, 190–195. [Google Scholar] [CrossRef] [PubMed]
- Patil, D.P.; Dhotre, D.P.; Chavan, S.G.; Sultan, A.; Jain, D.S.; Lanjekar, V.B.; Gangawani, J.; Shah, P.S.; Todkar, J.S.; Shah, S.; et al. Molecular analysis of gut microbiota in obesity among Indian individuals. J. Biosci. 2012, 37, 647–657. [Google Scholar] [CrossRef] [PubMed]
- Tims, S.; Derom, C.; Jonkers, D.M.; Vlietinck, R.; Saris, W.H.; Kleerebezem, M.; de Vos, W.M.; Zoetendal, E.G. Microbiota conservation and BMI signatures in adult monozygotic twins. ISME J. 2013, 7, 707–717. [Google Scholar] [CrossRef] [Green Version]
- Islam, M.M.; Lkhagva, E.; Chung, H.-J.; Hong, S.T. Development of a culture medium supporting universal growth of intestinal microbes. Nature, 2022; under review. [Google Scholar]
- Chung, H.-J.; Sharma, S.P.; Kim, H.-J.; Baek, S.-H.; Hong, S.T. The resveratrol-enriched rice DJ526 boosts motor coordination and physical strength. Sci. Rep. 2016, 6, 23958. [Google Scholar] [CrossRef] [Green Version]
- Baek, S.H.; Chung, H.-J.; Lee, H.K.; D’Souza, R.; Jeon, Y.; Kim, H.J.; Kweon, S.J.; Hong, S.T. Treatment of obesity with the resveratrol-enriched rice DJ526. Sci. Rep. 2014, 4, 3879. [Google Scholar] [CrossRef] [Green Version]
- Chung, H.-J.; Lee, H.K.; Kwon, K.B.; Kim, H.J.; Hong, S.T. Transferrin as a thermosensitizer in radiofrequency hyperthermia for cancer treatment. Sci. Rep. 2018, 8, e13505. [Google Scholar] [CrossRef]
- Lkhagva, E.; Chung, H.J.; Hong, J.; Tang, W.H.W.; Lee, S.I.; Hong, S.T.; Lee, S. The regional diversity of gut microbiome along the GI tract of male C57BL/6 mice. BMC Microbiol. 2021, 21, 44. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [Green Version]
- Ley, R.E.; Turnbaugh, P.J.; Klein, S.; Gordon, J.I. Microbial ecology. Human gut microbes associated with obesity. Nature 2006, 444, 1022–1023. [Google Scholar] [CrossRef]
- Jumpertz, R.; Le, D.S.; Turnbaugh, P.J.; Trinidad, C.; Bogardus, C.; Gordon, J.I.; Krakoff, J. Energy-balance studies reveal associations between gut microbes, caloric load, and nutrient absorption in humans. Am. J. Clin. Nutr. 2011, 94, 58–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bäckhed, F.; Ding, H.; Wang, T.; Hooper, L.V.; Koh, G.Y.; Nagy, A.; Semenkovich, C.F.; Gordon, J.I. The gut microbiota as an environmental factor that regulates fat storage. Proc. Natl. Acad. Sci. USA 2004, 101, 15718–15723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angelakis, E.; Armougom, F.; Million, M.; Raoult, D. The relationship between gut microbiota and weight gain in humans. Future Microbiol. 2012, 7, 91–109. [Google Scholar] [CrossRef] [PubMed]
- Turnbaugh, P.J.; Bäckhed, F.; Fulton, L.; Gordon, J.I. Diet-Induced Obesity Is Linked to Marked but Reversible Alterations in the Mouse Distal Gut Microbiome. Cell Host Microbe 2008, 3, 213–223. [Google Scholar] [CrossRef] [Green Version]
- Turnbaugh, P.J.; Ley, R.E.; Mahowald, M.A.; Magrini, V.; Mardis, E.R.; Gordon, J.I. Anobesity-associated gut microbiome with increased capacity for energy harvest. Nature 2006, 444, 1027–1031. [Google Scholar] [CrossRef]
- de Wit, N.; Derrien, M.; Bosch-Vermeulen, H.; Oosterink, E.; Keshtkar, S.; Duval, C.; de Vogel-van den Bosch, J.; Kleerebezem, M.; Müller, M.; van der Meer, R. Saturated fat stimulates obesity and hepatic steatosis and affects gut microbiota composition by an enhanced overflow of dietary fat to the distal intestine. Am. J. Physiol. Liver Physiol. 2012, 303, G589–G599. [Google Scholar] [CrossRef] [Green Version]
- De Filippo, C.; Cavalieri, D.; Di Paola, M.; Ramazzotti, M.; Poullet, J.B.; Massart, S.; Collini, S.; Pieraccini, G.; Lionetti, P. Impact of diet in shaping gut microbiota revealed by a comparative study in children from Europe and rural Africa. Proc. Natl. Acad. Sci. USA 2010, 107, 14691–14696. [Google Scholar] [CrossRef] [Green Version]
- Bervoets, L.; Van Hoorenbeeck, K.; Kortleven, I.; Van Noten, C.; Hens, N.; Vael, C.; Goossens, H.; Desager, K.N.; Vankerckhoven, V. Differences in gut microbiota composition between obese and lean children. A cross-sectional study. Gut Pathog. 2013, 5, 10. [Google Scholar] [CrossRef] [Green Version]
- Duan, M.; Wang, Y.; Zhang, Q.; Zou, R.; Guo, M.; Zheng, H. Characteristics of gut microbiota in people with obesity. PLoS ONE 2021, 16, e0255446. [Google Scholar] [CrossRef]
- Magne, F.; Gotteland, M.; Gauthier, L.; Zazueta, A.; Pesoa, S.; Navarrete, P.; Balamurugan, R. The Firmicutes/Bacteroidetes Ratio: A Relevant Marker of Gut Dysbiosis in Obese Patients? Nutrients 2020, 12, 1474. [Google Scholar] [CrossRef]
- Finucane, M.M.; Sharpton, T.J.; Laurent, T.J.; Pollard, K.S. A taxonomic signature of obesity in the microbiome? Getting to the guts of the matter. PLoS ONE 2014, 9, e84689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sze, M.A.; Schloss, P.D. Looking for a Signal in the Noise. Revisiting Obesity and the Microbiome. mBio 2016, 7, e01018-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhai, C.; Ahn, J.-S.; Islam, M.M.; Lkhagva, E.; Chung, H.-J.; Hong, S.-T. Comparative Analysis of Original and Replaced Gut Microbiomes within Same Individuals Identified the Intestinal Microbes Associated with Weight Gaining. Microorganisms 2022, 10, 1062. https://doi.org/10.3390/microorganisms10051062
Zhai C, Ahn J-S, Islam MM, Lkhagva E, Chung H-J, Hong S-T. Comparative Analysis of Original and Replaced Gut Microbiomes within Same Individuals Identified the Intestinal Microbes Associated with Weight Gaining. Microorganisms. 2022; 10(5):1062. https://doi.org/10.3390/microorganisms10051062
Chicago/Turabian StyleZhai, Chongkai, Ji-Seon Ahn, Md Minarul Islam, Enkhchimeg Lkhagva, Hea-Jong Chung, and Seong-Tshool Hong. 2022. "Comparative Analysis of Original and Replaced Gut Microbiomes within Same Individuals Identified the Intestinal Microbes Associated with Weight Gaining" Microorganisms 10, no. 5: 1062. https://doi.org/10.3390/microorganisms10051062