
Genomic Diversity of SARS-CoV-2 in Algeria and North African Countries: What We Know So Far and What We Expect?


Abstract
:1. Introduction
2. Materials and Methods
2.1. Epidemiological Dynamics and Genomic Data Processing
2.2. Sequence Alignment and Phylogenetic Analysis of Algerian Genomes
2.3. Mutation Signature and Clade Assignment Analysis
3. Results
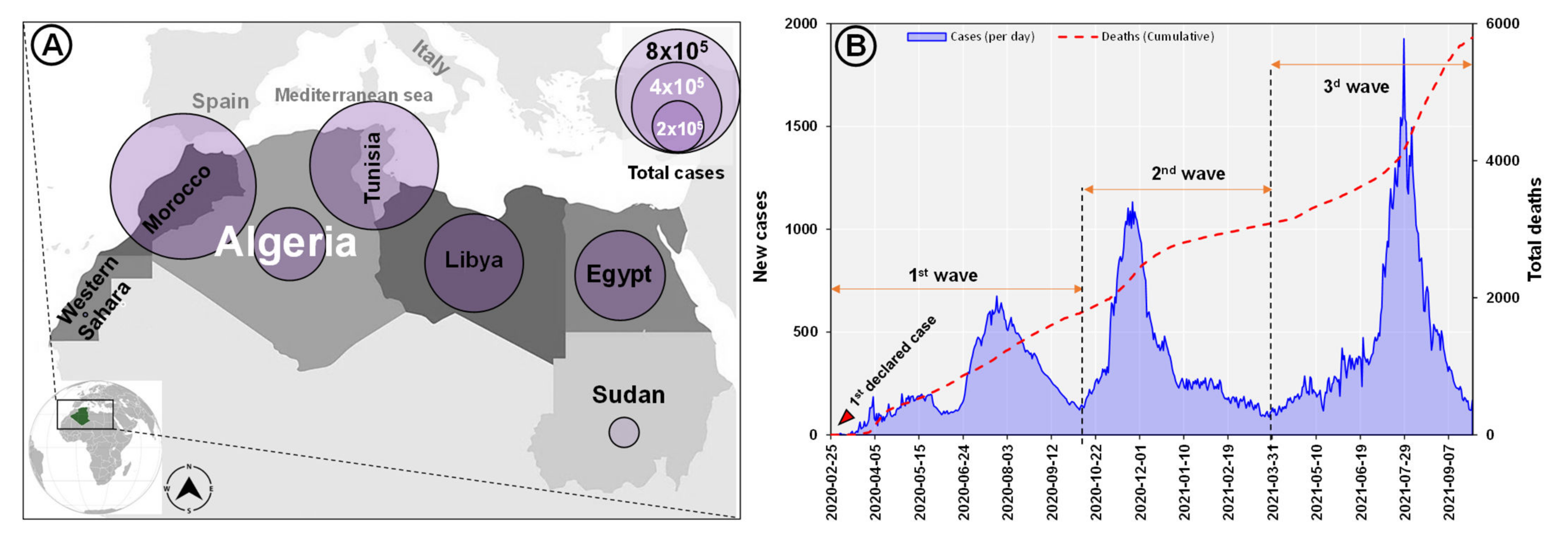
3.1. Epidemiology of SARS-CoV-2 in Algeria and North Africa
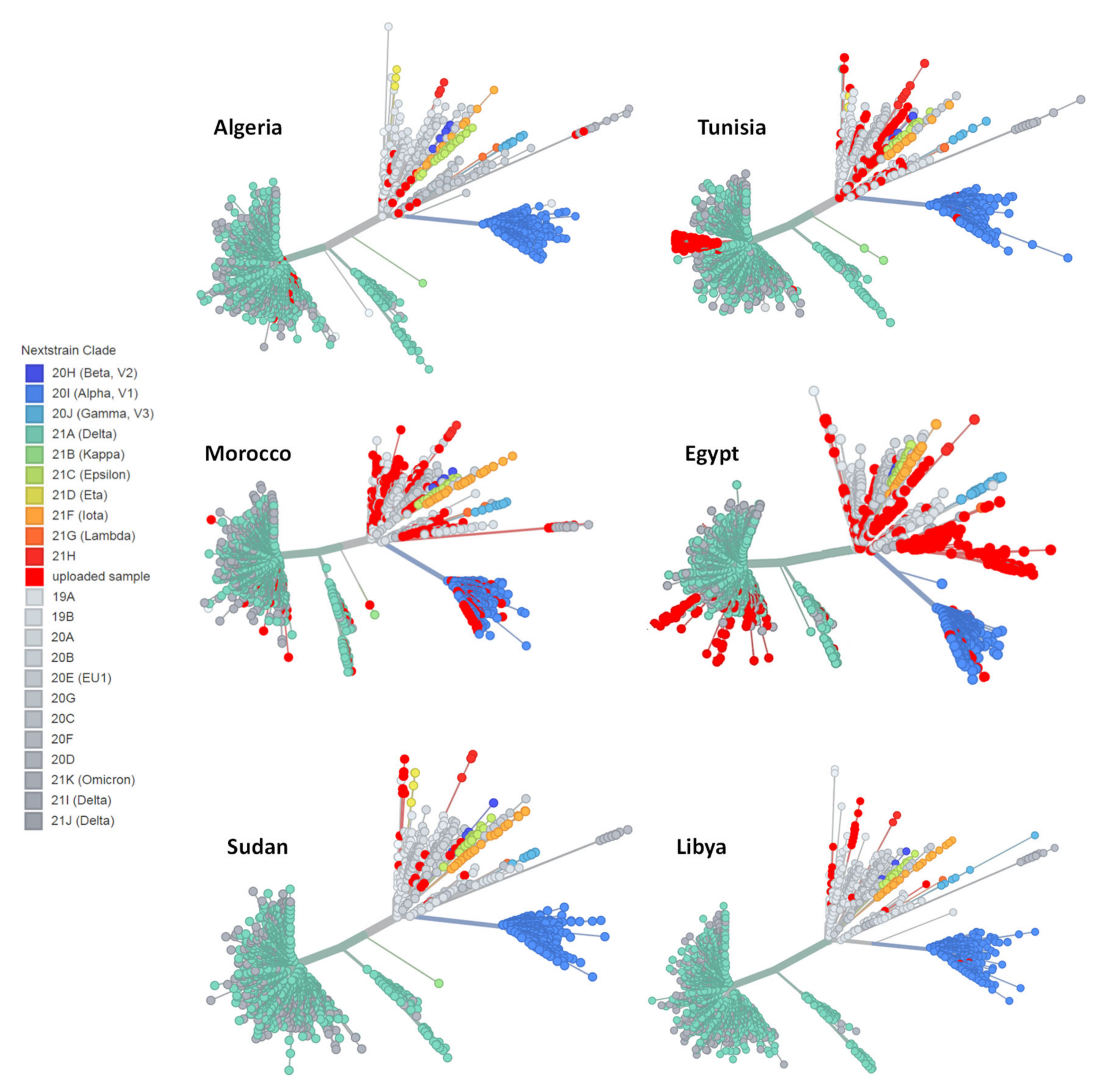
3.2. Phylogenetic Analysis of SARS-CoV-2 Genomes in Algeria
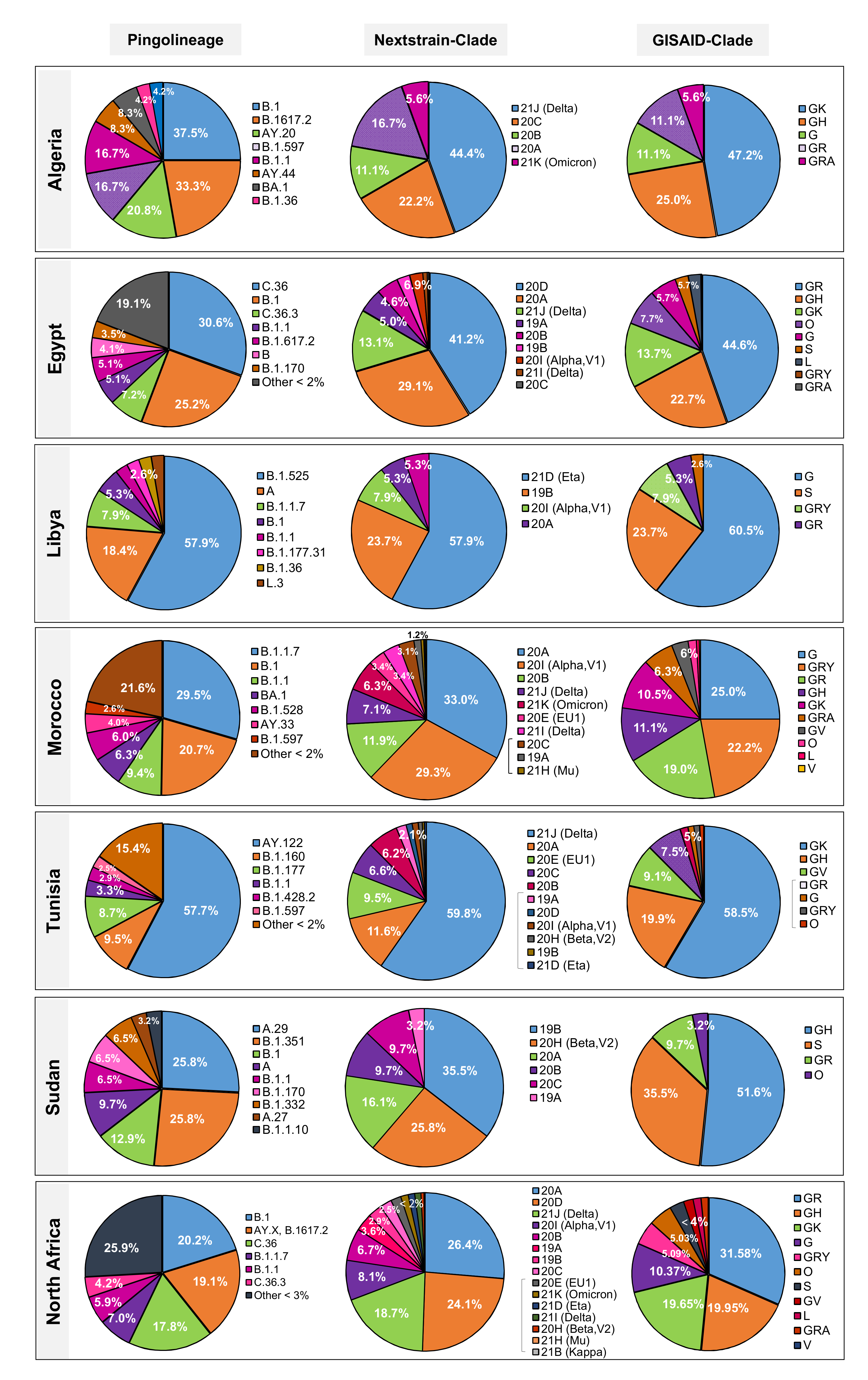
3.3. Distribution of SARS-CoV-2 Lineages and Clades in North Africa
3.3.1. Egypt
3.3.2. Libya
3.3.3. Morocco
3.3.4. Sudan
3.3.5. Tunisia
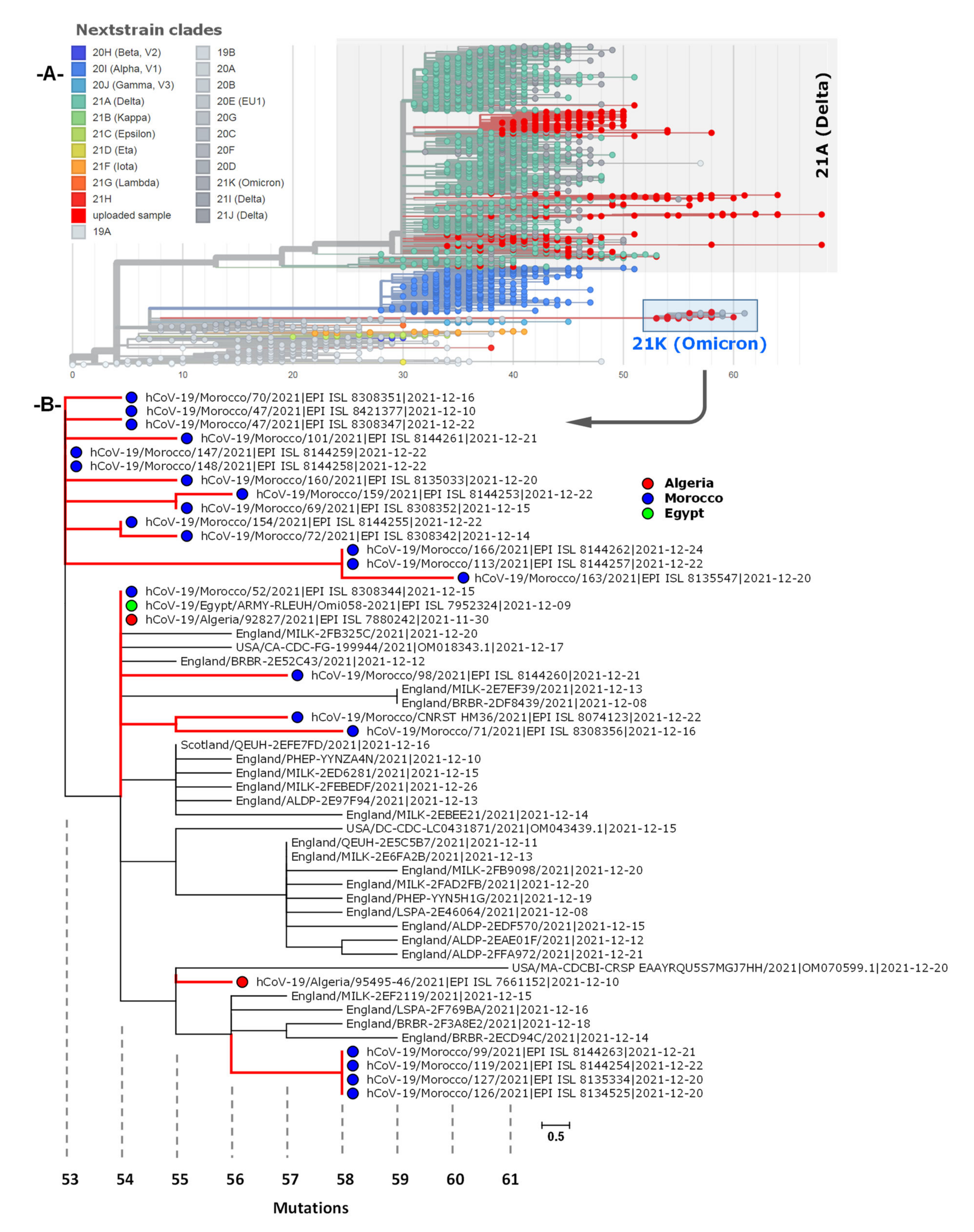
3.4. Phylogenetic Analysis of Omicron Genome Sequences from North Africa
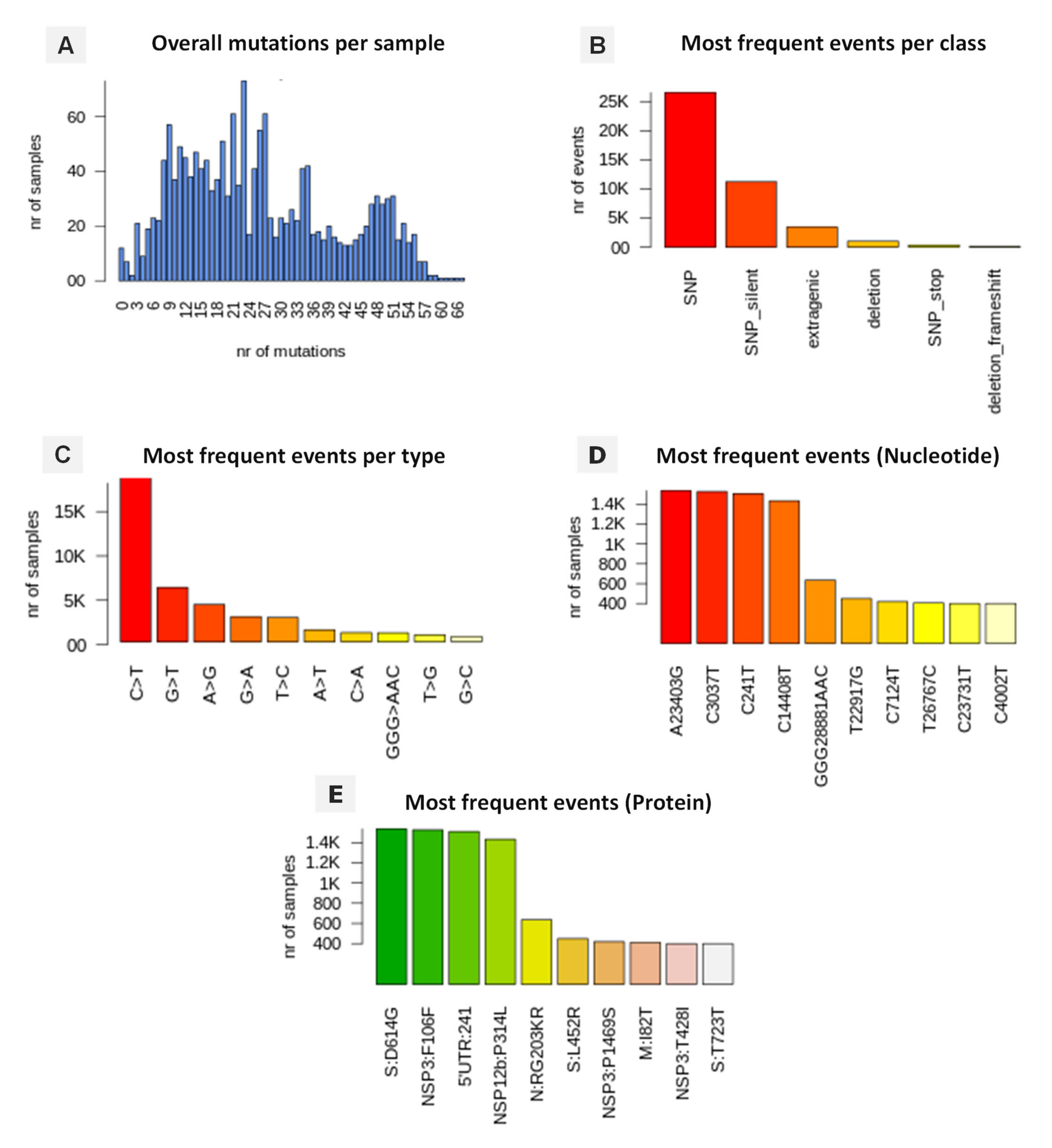
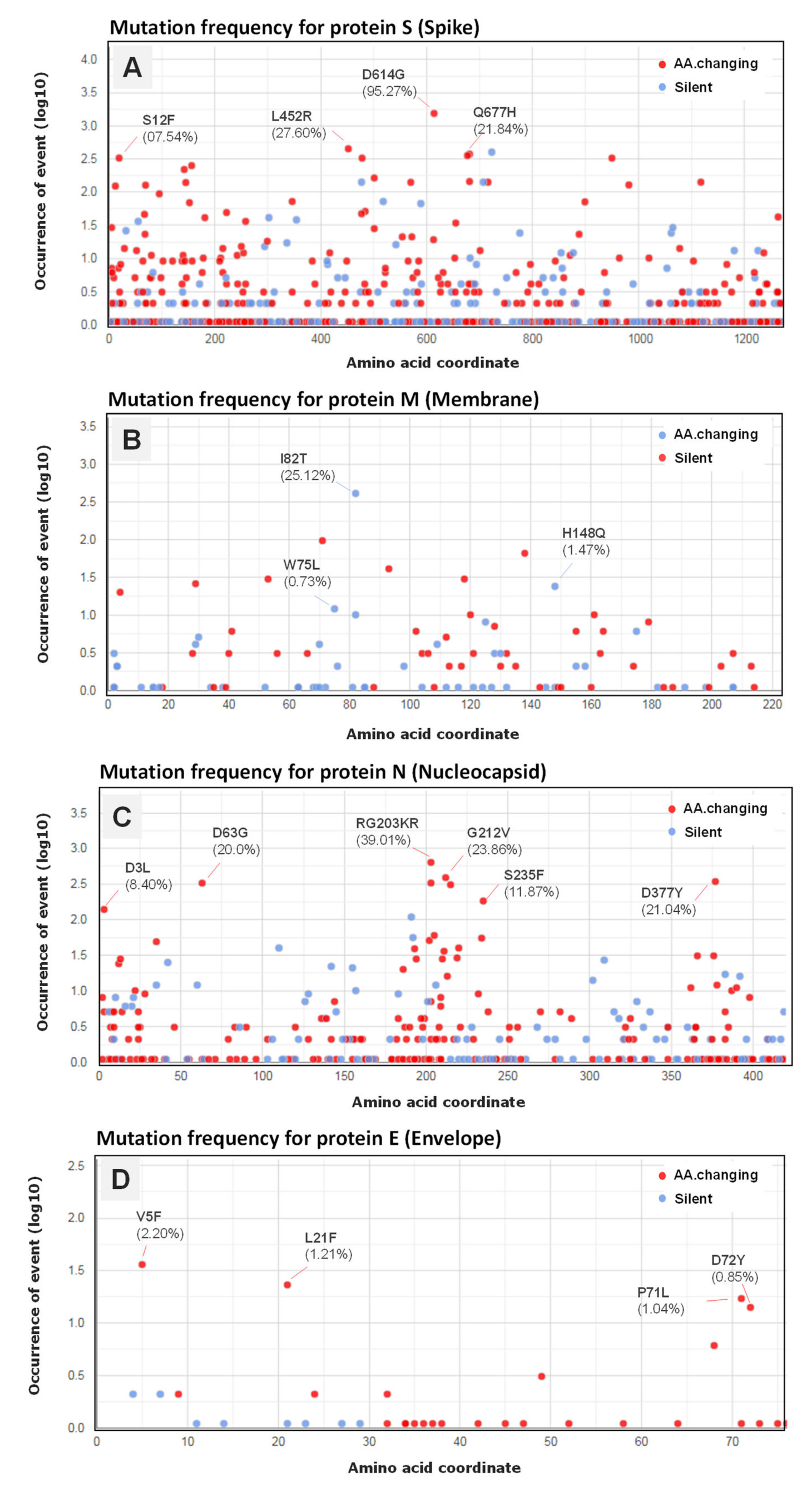
3.5. Genomic Variation and Mutation Signature
3.6. Variant Analysis of Omicron SARS-CoV-2 Genomes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hui, D.S.; Azhar, E.I.; Madani, T.A.; Ntoumi, F.; Kock, R.; Dar, O.; Ippolito, G.; McHugh, T.D.; Memish, Z.A.; Drosten, C.; et al. The continuing 2019-nCoV epidemic threat of novel coronaviruses to global health—The latest 2019 novel coronavirus outbreak in Wuhan, China. Int. J. Infect. Dis. 2020, 91, 264–266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- WHO. Novel Coronavirus–China. 2020. Available online: https://www.who.int/csr/don/12-january-2020-novel-coronavirus-china/en/ (accessed on 27 August 2021).
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.M.; Wang, W.; Song, Z.G.; Hu, Y.; Tao, Z.-W.; Tian, J.-H.; Pei, Y.-Y.; et al. A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, J.F.W.; Yuan, S.; Kok, K.H.; To, K.K.W.; Chu, H.; Yang, J.; Xing, F.; Liu, J.; Yip, C.C.-Y.; Poon, R.W.-S.; et al. A familial cluster of pneumonia associated with the 2019 novel coronavirus indicating person-to-person transmission: A study of a family cluster. Lancet 2020, 395, 514–523. [Google Scholar] [CrossRef] [Green Version]
- Khosrawipour, V.; Lau, H.; Khosrawipour, T.; Kocbach, P.; Ichii, H.; Bania, J.; Mikolajczyk, A. Failure in initial stage containment of global COVID-19 epicenters. J. Med. Virol. 2020, 92, 863–867. [Google Scholar] [CrossRef] [Green Version]
- WHO. Coronavirus Disease 2019 (COVID-19). Weekly Epidemiological Update on COVID-19. 21 December 2021. Available online: https://www.who.int/publications/m/item/weekly-epidemiological-update-on-covid-19---21- (accessed on 21 December 2021).
- Van Woensel, J.B.M.; Van Aalderen, W.M.C.; Kimpen, J.L.L. Viral lower respiratory tract infection in infants and young children. BMJ 2003, 327, 36–40. [Google Scholar] [CrossRef] [Green Version]
- Grant, R.; Malik, M.R.; Elkholy, A.; Van Kerkhove, M.D. A review of asymptomatic and sub-clinical Middle East Respiratory Syndrome Coronavirus Infections. Epidemiol. Rev. 2019, 41, 69–81. [Google Scholar] [CrossRef] [Green Version]
- Azhar, E.I.; El-Kafrawy, S.A.; Farraj, S.A.; Hassan, A.M.; Al-Saeed, M.S.; Hashem, A.M.; Madani, T.A. Evidence for camel-to-human transmission of MERS coronavirus. N. Engl. J. Med. 2014, 370, 2499–2505. [Google Scholar] [CrossRef]
- Zumla, A.; Memish, Z.A.; Hui, D.S.; Perlman, S. Vaccine against Middle East respiratory syndrome coronavirus. Lancet Infect. Dis. 2019, 19, 1054–1055. [Google Scholar] [CrossRef] [Green Version]
- WHO. Coronavirus Disease 2019 (COVID-19). Situation Report–37. Data as Reported as 27 February 2020. 2020. Available online: https://www.who.int/docs/default-source/coronaviruse/situation-reports/20200226-sitrep-37-covid-19.pdf?sfvrsn=2146841e_2 (accessed on 25 December 2021).
- AFRICACDC. 2020. Available online: https://africacdc.org/download/covid-19-scientific-and-public-health-policy-update-17-august-2021/ (accessed on 25 December 2021).
- Janik, E.; Niemcewicz, M.; Podogrocki, M.; Majsterek, I.; Bijak, M. The Emerging Concern and Interest SARS-CoV-2 Variants. Pathogens 2021, 10, 633. [Google Scholar] [CrossRef]
- Rella, S.A.; Kulikova, Y.A.; Dermitzakis, E.T.; Kondrashov, F.A. Rates of SARS-CoV-2 transmission and vaccination impact the fate of vaccine-resistant strains. Sci. Rep. 2021, 11, 1–10. [Google Scholar] [CrossRef]
- WHO. Tracking SARS-CoV-2 Variants. 2021. Available online: https://www.who.int/en/activities/tracking-SARS-CoV-2-variants/ (accessed on 30 December 2021).
- Shu, Y.; McCauley, J. GISAID: Global initiative on sharing all influenza data–from vision to reality. Eurosurveillance 2017, 22, 30494. [Google Scholar] [CrossRef] [Green Version]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; Von Haeseler, A.; Lanfear, R. IQ-TREE 2: New models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef] [Green Version]
- Rambaut, A.; Loman, N.; Pybus, O.; Barclay, W.; Barrett, J.; Carabell, A.; Connor, T.; Peacock, T.; Robertson, D.L.; Volz, E.; et al. Preliminary Genomic Characterisation of an Emergent SARS-CoV-2 Lineage in the UK Defined by a Novel Set of Spike Mutations. Available online: https://virological.org/t/preliminary-genomic-characterisation-of-an-emergent-sars-cov-2-lineage-in-the-uk-defined-by-a-novel-set-of-spike-mutations/563 (accessed on 20 December 2021).
- Hadfield, J.; Megill, C.; Bell, S.M.; Huddleston, J.; Potter, B.; Callenderet, C.; Sagulenko, P.; Bedford, T.; Neher, R.A. Nextstrain: Real-time tracking of pathogen evolution. Bioinformatics 2018, 34, 4121–4123. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [Green Version]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v4: Recent updates and new developments. Nucleic Acids Res. 2019, 47, W256–W259. [Google Scholar] [CrossRef] [Green Version]
- Mercatelli, D.; Giorgi, F.M. Geographic and genomic distribution of SARS-CoV-2 mutations. Front. Microbiol. 2020, 11, 1800. [Google Scholar] [CrossRef]
- MSPRH, Plan de Préparation et de Riposte à la Menace de L’infection Coronavirus COVID-19. 2020. Available online: https://www.sante.gov.dz/coronavirus/coronavirus-2019.html (accessed on 10 December 2021).
- WHO. COVID-19 Strategic Response Plan in the WHO African Region. 2020. Available online: https://www.afro.who.int/publications/covid-19-strategic-response-plan-who-african-region (accessed on 20 December 2021).
- Impouma, B.; Mboussou, F.; Wolfe, C.M.; Farham, B.; Williams, G.S.; Ogundiran, O.; Ngom, R.; Nzingou, M.; Flahault, A.; Codeço, C.T.; et al. COVID-19 in the WHO African Region: Using risk assessment to inform decisions on public health and social measures. Epidemiol. Infect. 2021, 149, 1–22. [Google Scholar] [CrossRef]
- Schröder, M.; Bossert, A.; Kersting, M.; Aeffner, S.; Coetzee, J.; Timme, M.; Schlüter, J. COVID-19 in South Africa: Outbreak despite interventions. Sci. Rep. 2021, 11, 1–9. [Google Scholar] [CrossRef]
- Benvenuto, D.; Giovanetti, M.; Salemi, M.; Prosperi, M.; De Flora, C.; Junior Alcantara, L.C.; Angeletti, S.; Ciccozzi, M. The global spread of 2019-nCoV: A molecular evolutionary analysis. Pathog. Glob. Health 2020, 114, 64–67. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez-Reiche, A.S.; Hernandez, M.M.; Sullivan, M.J.; Ciferri, B.; Alshammary, H.; Obla, A.; Fabre, S.; Kleiner, G.; Polanco, J.; Khan, Z.; et al. Introductions and early spread of SARS-CoV-2 in the New York City area. Science 2020, 369, 297–301. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Wu, C.; Li, X.; Song, Y.; Yao, X.; Wu, X.; Duan, Y.; Zhang, H.; Wang, Y.; Qian, Z.; et al. On the origin and continuing evolution of SARS-CoV-2. Nat. Sci. Rev. 2020, 7, 1012–1023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamed, S.M.; Elkhatib, W.F.; Khairalla, A.S.; Noreddin, A.M. Global dynamics of SARS-CoV-2 clades and their relation to COVID-19 epidemiology. Sci. Rep. 2021, 11, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Omais, S.O.; Kharroubi, S.; Zaraket, H. No association between the SARS-CoV-2 variants and mortality rates in the Eastern Mediterranean Region. medRxiv 2021. [Google Scholar] [CrossRef]
- Zekri, A.R.N.; Amer, K.E.; Hafez, M.M.; Hassan, Z.K.; Ahmed, O.S.; Soliman, H.K.; Bahnasy, A.A.; Hamid, W.A.; Gad, A.; Ali, M.; et al. Genomic characterization of SARS-CoV-2 in Egypt. J. Adv. Res. 2021, 30, 123–132. [Google Scholar] [CrossRef]
- Badaoui, B.; Sadki, K.; Talbi, C.; Salah, D.; Tazi, L. Genetic diversity and genomic epidemiology of SARS-CoV-2 in Morocco. Biosaf. Health 2021, 3, 124–127. [Google Scholar] [CrossRef]
- Faria, N.R.; Mellan, T.A.; Whittaker, C.; Claro, I.M.; Candido, D.D.S.; Mishra, S.; Crispim, M.A.E.; Sales, F.C.S.; Hawryluk, I.; McCrone, J.T.; et al. Genomics and epidemiology of the P. 1 SARS-CoV-2 lineage in Manaus, Brazil. Science 2021, 372, 815–821. [Google Scholar] [CrossRef]
- Voloch, C.M.; da Silva Francisco, R., Jr.; de Almeida, L.G.; Cardoso, C.C.; Brustolini, O.J.; Gerber, A.L.; Guimarães, A.P.D.C.; Mariani, D.; da Costa, R.M.; Ferreira, O.C.; et al. Genomic characterization of a novel SARS-CoV-2 lineage from Rio de Janeiro, Brazil. J. Virol. 2021, 95, e00119–e00121. [Google Scholar] [CrossRef]
- Tegally, H.; Wilkinson, E.; Giovanetti, M.; Iranzadeh, A.; Fonseca, V.; Giandhari, J.; Doolabh, D.; Pillay, S.; San, E.J.; Msomi, N.; et al. Detection of a SARS-CoV-2 variant of concern in South Africa. Nature 2021, 592, 438–443. [Google Scholar] [CrossRef]
- Yadav, P.; Sapkal, G.N.; Abraham, P.; Ella, R.; Deshpande, G.; Patil, D.; Nyayanit, D.A.; Gupta, N.; Sahay, R.R.; Shete, A.M.; et al. Neutralization of variant under investigation B. 1.617 with sera of BBV152 vaccinees. Clin. Infect. Dis. 2021, 74, 366–368. [Google Scholar] [CrossRef]
- He, X.; Hong, W.; Pan, X.; Lu, G.; Wei, X. SARS-CoV-2 Omicron variant: Characteristics and prevention. MedComm 2021, 2, 838–845. [Google Scholar] [CrossRef]
- Gobeil, S.M.C.; Janowska, K.; McDowell, S.; Mansouri, K.; Parks, R.; Manne, K.; Stalls, V.; Kopp, M.F.; Henderson, R.; Edwards, R.J.; et al. D614G mutation alters SARS-CoV-2 spike conformation and enhances protease cleavage at the S1/S2 junction. Cell Rep. 2021, 34, 108630. [Google Scholar] [CrossRef]
- Korber, B.; Fischer, W.M.; Gnanakaran, S.; Yoon, H.; Theiler, J.; Abfalterer, W.; Hengartner, N.; Giorgi, E.E.; Bhattacharya, T.; Foley, B.; et al. Tracking changes in SARS-CoV-2 spike: Evidence that D614G increases infectivity of the COVID-19 virus. Cell 2020, 182, 812–827. [Google Scholar] [CrossRef]
- Ogawa, J.; Zhu, W.; Tonnu, N.; Singer, O.; Hunter, T.; Ryan, A.L.; Pao, G.M. The D614G mutation in the SARS-CoV2 Spike protein increases infectivity in an ACE2 receptor dependent manner. Biorxiv 2020, 22, 214932. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.-H.; Nitsche, A.; et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 2020, 181, 271–280. [Google Scholar] [CrossRef]
- Lan, J.; Ge, J.; Yu, J.; Shan, S.; Zhou, H.; Fan, S.; Zhang, Q.; Shi, X.; Wang, Q.; Zhang, L.; et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 2020, 581, 215–220. [Google Scholar] [CrossRef] [Green Version]
- McAuley, A.J.; Kuiper, M.J.; Durr, P.A.; Bruce, M.P.; Barr, J.; Todd, S.; Au, G.G.; Blasdell, K.; Tachedjian, M.; Lowther, S.; et al. Experimental and in silico evidence suggests vaccines are unlikely to be affected by D614G mutation in SARS-CoV-2 spike protein. NPJ Vaccines 2020, 5, 1–5. [Google Scholar] [CrossRef]
- Yalcin, H.C.; Sukumaran, V.; Al-Ruweidi, M.K.A.; Shurbaji, S. Do changes in ace-2 expression affect SARS-CoV-2 virulence and related complications: A closer look into membrane-bound and soluble forms. Int. J. Mol. Sci. 2021, 22, 6703. [Google Scholar] [CrossRef]
- Yin, C. Genotyping coronavirus SARS-CoV-2: Methods and implications. Genomics 2020, 112, 3588–3596. [Google Scholar] [CrossRef]
- Naveca, F.; da Costa, C.; Nascimento, V.; Souza, V.; Corado, A.; Nascimento, F.; Costa, Á.; Duarte, D.; Silva, G.; Mejía, M.; et al. SARS-CoV-2 Reinfection by the New Variant of Concern (VOC) P.1 in Amazonas, Brazil. 2021. Available online: https://virological.org/t/sars-cov-2-reinfection-by-the-new-variant-of-concern-voc-p-1-in-amazonas-brazil/596 (accessed on 30 December 2021).
- McCarthy, K.R.; Rennick, L.J.; Nambulli, S.; Robinson-McCarthy, L.R.; Bain, W.G.; Haidar, G.; Duprex, W.P. Recurrent deletions in the SARS-CoV-2 spike glycoprotein drive antibody escape. Science 2021, 71, 1139–1142. [Google Scholar] [CrossRef]
- Garcia-Beltran, W.F.; Lam, E.C.; Denis, K.S.; Nitido, A.D.; Garcia, Z.H.; Hauser, B.M.; Feldman, J.; Pavlovic, M.N.; Gregory, D.J.; Poznansky, M.C.; et al. Multiple SARS-CoV-2 variants escape neutralization by vaccine-induced humoral immunity. Cell 2021, 184, 2372–2383. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Liu, J.; Zhou, X. A CRISPR-based and post-amplification coupled SARS-CoV-2 detection with a portable evanescent wave biosensor. Biosens. Bioelectron. 2021, 190, 113418. [Google Scholar] [CrossRef]
- Zhu, Q.; Zhou, X. A colorimetric sandwich-type bioassay for SARS-CoV-2 using a hACE2-based affinity peptide pair. J. Hazard. Mater. 2022, 425, 127923. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J. Omicron sparks a vaccine strategy debate. Science 2021, 374, 1544–1545. [Google Scholar] [CrossRef] [PubMed]
- Karanikolos, M.; McKee, M. How comparable is COVID-19 mortality across countries? Eurohealth 2020, 26, 45–50. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Country | Total Cases | Sequenced Genomes | Analyzed Genomes |
|---|---|---|---|
| Algeria | 214,592 | 85 | 36 |
| Egypt | 375,330 | 1418 | 971 |
| Libya | 381,749 | 56 | 38 |
| Morocco | 952,814 | 609 | 352 |
| Sudan | 45,112 | 116 | 31 |
| Tunisia | 721,031 | 315 | 241 |
| Western Sahara | 10 | ND | ND |
| Total | 2,690,638 | 2599 | 1669 |
| Genomic Coordinate | Effect | N Samples | Class | Genomic Region |
|---|---|---|---|---|
| A23403G | S:D614G | 25 | SNP | Spike protein |
| G23948T | S:D796Y | 25 | ||
| C23202A | S:T547K | 25 | ||
| C24130A | S:N856K | 25 | ||
| T23599G | S:N679K | 25 | ||
| T24469A | S:N969K | 25 | ||
| C23604A | S:P681H | 25 | ||
| C24503T | S:L981F | 25 | ||
| A24424T | S:Q954H | 24 | ||
| C23854A | S:N764K | 24 | ||
| C23525T | S:H655Y | 24 | ||
| G22813T | S:K417N | 24 | ||
| G22578A | S:G339D | 24 | ||
| T22195G | S:N211K | 23 | ||
| A23040G | S:Q493R | 22 | ||
| TC22673CT | S:S371L | 22 | ||
| T22679C | S:S373P | 22 | ||
| C22686T | S:S375F | 22 | ||
| G22992A | S:S477N | 22 | ||
| C22995A | S:T478K | 22 | ||
| A23013C | S:E484A | 22 | ||
| G22898A | S:G446S | 21 | ||
| C10029T | NSP4:T492I | 25 | SNP SNP | Transmembrane protein |
| A11537G | NSP6:I189V | 25 | ||
| 11286TGTCTGGTT | NSP6:L105 | 24 | Deletion | |
| G8393A | NSP3:A1892T | 25 | SNP | Predicted phosphoesterase |
| A2832G | NSP3:K38R | 24 | SNP | |
| 6513GTT | NSP3:S1265 | 24 | Deletion | |
| C28311T | N:P13L | 24 | SNP | Nucleocapsid protein |
| GGG2881AAC | N:RG203KR | 24 | ||
| G26709A | M:A63T | 24 | SNP | Membrane |
| A26530G | M:D3G | 22 | ||
| C26577G | M:Q19E | 24 | ||
| C26270T | E:T9I | 25 | SNP | Envelope |
| A18163G | NSP14:I42V | 22 | SNP | 3′-to-5′ exonuclease |
| C10449A | NSP5:P132H | 24 | SNP | 3C-like proteinase |
| C14408T | NSP12b:P314L | 25 | SNP | RNA-dependent RNA polymerase |
| A28271T | 3′UTR:28271 | 25 | Extragenic | 3′ Untranslated Region |
| C241T | 5′UTR:241 | 24 | Extragenic | 5′ Untranslated Region |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Menasria, T.; Aguilera, M. Genomic Diversity of SARS-CoV-2 in Algeria and North African Countries: What We Know So Far and What We Expect? Microorganisms 2022, 10, 467. https://doi.org/10.3390/microorganisms10020467
Menasria T, Aguilera M. Genomic Diversity of SARS-CoV-2 in Algeria and North African Countries: What We Know So Far and What We Expect? Microorganisms. 2022; 10(2):467. https://doi.org/10.3390/microorganisms10020467
Chicago/Turabian StyleMenasria, Taha, and Margarita Aguilera. 2022. "Genomic Diversity of SARS-CoV-2 in Algeria and North African Countries: What We Know So Far and What We Expect?" Microorganisms 10, no. 2: 467. https://doi.org/10.3390/microorganisms10020467