
Spread of a Novel Indian Ocean Lineage Carrying E1-K211E/E2-V264A of Chikungunya Virus East/Central/South African Genotype across the Indian Subcontinent, Southeast Asia, and Eastern Africa

 ,
,  , , , ,
, , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. CHIKV Samples
2.2. CHIKV Genome Sequencing
2.3. Data Collection and Phylogenetics
2.4. Selection Analysis
3. Results
3.1. CHIKV Obtained in the Present Study
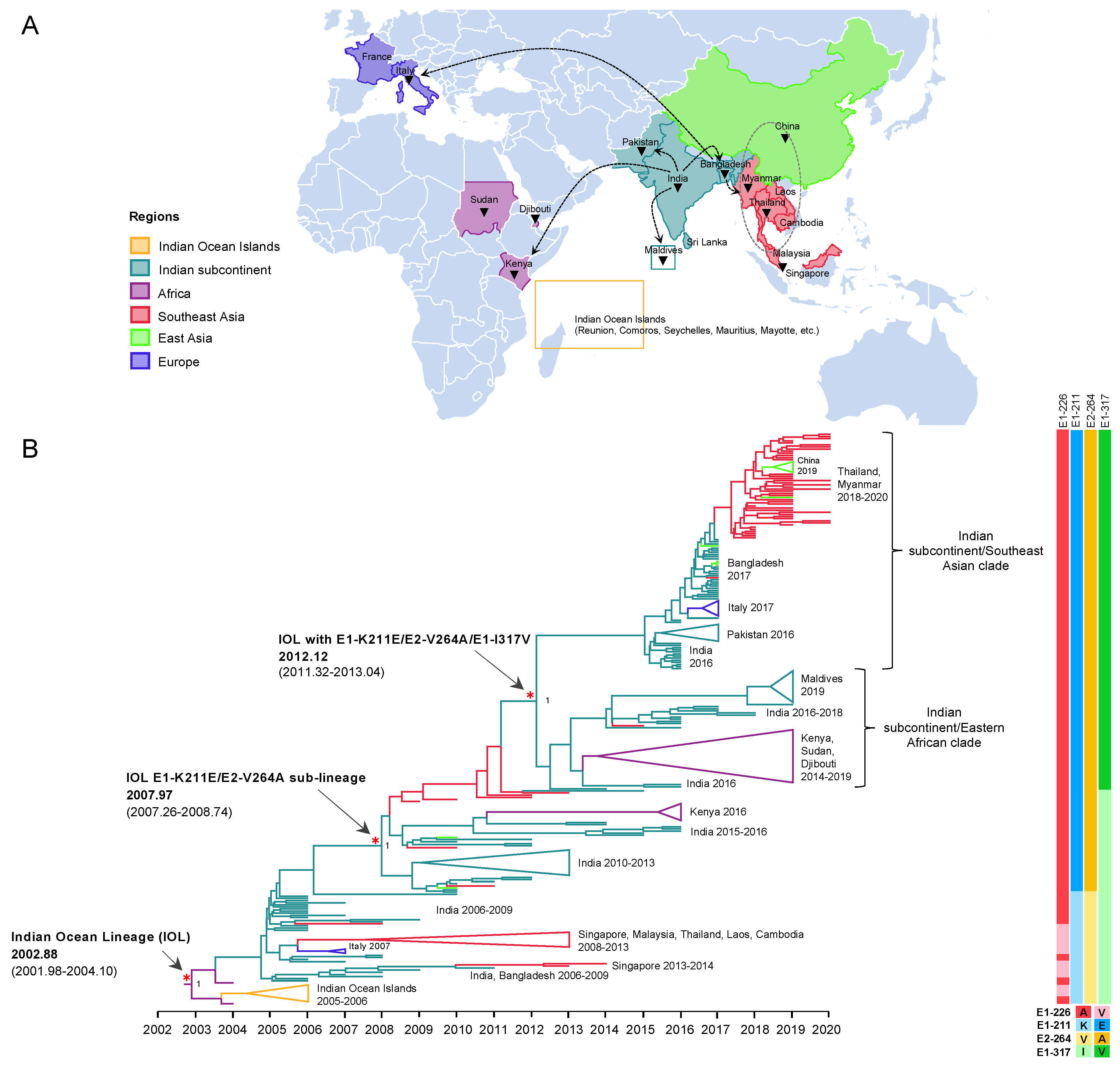
3.2. Distribution of Indian Ocean Lineage Carrying E1-K211E and E2-V264A of CHIKV ECSA
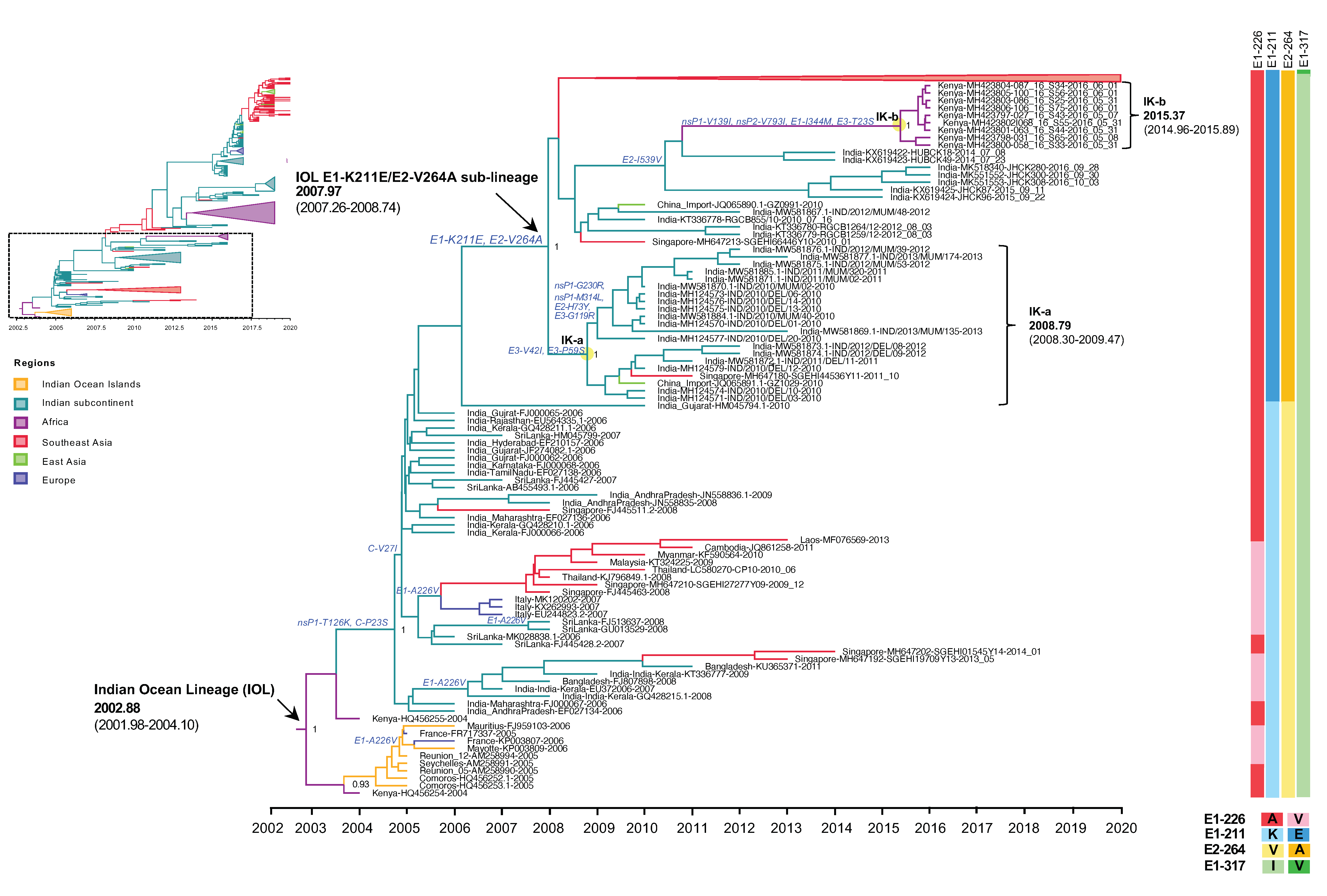
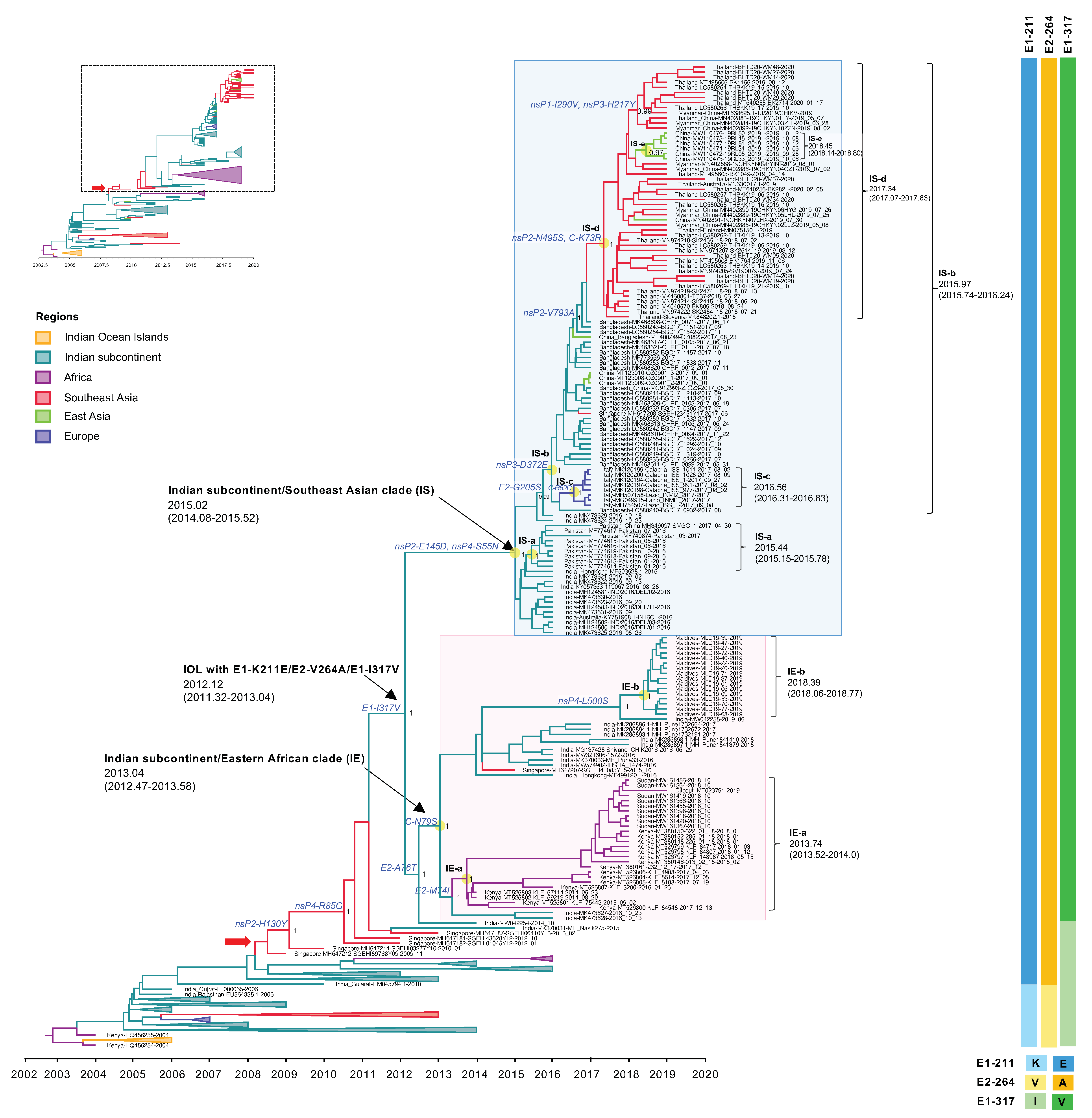
3.3. Evolutionary Dynamics of the Indian Ocean Lineage of CHIKV ECSA
3.3.1. The Emergence of IOL Sub-Lineage E1-K211E/E2-V264A
3.3.2. The Expansion of IOL Sub-Lineage E1-K211E/E2-V264A
3.4. Selection Analyses
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Volk, S.M.; Chen, R.; Tsetsarkin, K.A.; Adams, A.P.; Garcia, T.I.; Sall, A.A.; Nasar, F.; Schuh, A.J.; Holmes, E.C.; Higgs, S.; et al. Genome-scale phylogenetic analyses of chikungunya virus reveal independent emergences of recent epidemics and various evolutionary rates. J. Virol. 2010, 84, 6497–6504. [Google Scholar] [CrossRef] [Green Version]
- Weaver, S.C.; Chen, R.; Diallo, M. Chikungunya Virus: Role of Vectors in Emergence from Enzootic Cycles. Annu. Rev. Entomol. 2020, 65, 313–332. [Google Scholar] [CrossRef] [Green Version]
- Tsetsarkin, K.A.; Vanlandingham, D.L.; McGee, C.E.; Higgs, S. A single mutation in chikungunya virus affects vector specificity and epidemic potential. PLoS Pathog. 2007, 3, e201. [Google Scholar] [CrossRef]
- Shi, J.; Su, Z.; Fan, Z.; Wang, J.; Liu, S.; Zhang, B.; Wei, H.; Jehan, S.; Jamil, N.; Shen, S.; et al. Extensive evolution analysis of the global chikungunya virus strains revealed the origination of CHIKV epidemics in Pakistan in 2016. Virol. Sin. 2017, 32, 520–532. [Google Scholar] [CrossRef]
- Agarwal, A.; Sharma, A.K.; Sukumaran, D.; Parida, M.; Dash, P.K. Two novel epistatic mutations (E1:K211E and E2:V264A) in structural proteins of Chikungunya virus enhance fitness in Aedes aegypti. Virology 2016, 497, 59–68. [Google Scholar] [CrossRef]
- Phadungsombat, J.; Imad, H.; Rahman, M.; Nakayama, E.E.; Kludkleeb, S.; Ponam, T.; Rahim, R.; Hasan, A.; Poltep, K.; Yamanaka, A.; et al. A Novel Sub-Lineage of Chikungunya Virus East/Central/South African Genotype Indian Ocean Lineage Caused Sequential Outbreaks in Bangladesh and Thailand. Viruses 2020, 12, 1319. [Google Scholar] [CrossRef]
- Venturi, G.; Di Luca, M.; Fortuna, C.; Remoli, M.E.; Riccardo, F.; Severini, F.; Toma, L.; Del Manso, M.; Benedetti, E.; Caporali, M.G.; et al. Detection of a chikungunya outbreak in Central Italy, August to September 2017. Eurosurveillance 2017, 22, 17-00646. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.B.; Li, M.; Gao, N.; Shen, J.Y.; Sheng, Z.Y.; Fan, D.Y.; Zhou, H.N.; Yin, X.X.; Mao, J.R.; Jiang, J.Y.; et al. Epidemiological and Clinical Characteristics of the Chikungunya Outbreak in Ruili City, Yunnan Province, China. J. Med. Virol. 2021, 94, 499–506. [Google Scholar] [CrossRef]
- Fourie, T.; Dia, A.; Savreux, Q.; Pommier de Santi, V.; de Lamballerie, X.; Leparc-Goffart, I.; Simon, F. Emergence of Indian lineage of ECSA chikungunya virus in Djibouti, 2019. Int. J. Infect. Dis. 2021, 108, 198–201. [Google Scholar] [CrossRef]
- Bower, H.; El Karsany, M.; Adam, A.; Idriss, M.I.; Alzain, M.A.; Alfakiyousif, M.E.A.; Mohamed, R.; Mahmoud, I.; Albadri, O.; Mahmoud, S.A.A.; et al. “Kankasha” in Kassala: A prospective observational cohort study of the clinical characteristics, epidemiology, genetic origin, and chronic impact of the 2018 epidemic of Chikungunya virus infection in Kassala, Sudan. PLoS Negl. Trop. Dis. 2021, 15, e0009387. [Google Scholar] [CrossRef]
- Nyamwaya, D.K.; Otiende, M.; Omuoyo, D.O.; Githinji, G.; Karanja, H.K.; Gitonga, J.N.; Laurent, Z.R.d.; Otieno, J.R.; Sang, R.; Kamau, E.; et al. Endemic chikungunya fever in Kenyan children: A prospective cohort study. BMC Infect. Dis. 2021, 21, 186. [Google Scholar] [CrossRef] [PubMed]
- Maljkovic Berry, I.; Eyase, F.; Pollett, S.; Konongoi, S.L.; Joyce, M.G.; Figueroa, K.; Ofula, V.; Koka, H.; Koskei, E.; Nyunja, A.; et al. Global Outbreaks and Origins of a Chikungunya Virus Variant Carrying Mutations Which May Increase Fitness for Aedes aegypti: Revelations from the 2016 Mandera, Kenya Outbreak. Am. J. Trop. Med. Hyg. 2019, 100, 1249–1257. [Google Scholar] [CrossRef] [PubMed]
- Pickett, B.E.; Sadat, E.L.; Zhang, Y.; Noronha, J.M.; Squires, R.B.; Hunt, V.; Liu, M.; Kumar, S.; Zaremba, S.; Gu, Z.; et al. ViPR: An open bioinformatics database and analysis resource for virology research. Nucleic Acids Res. 2012, 40, D593–D598. [Google Scholar] [CrossRef]
- Imad, H.A.; Phadungsombat, J.; Nakayama, E.E.; Suzuki, K.; Ibrahim, A.M.; Afaa, A.; Azeema, A.; Nazfa, A.; Yazfa, A.; Ahmed, A.; et al. Clinical Features of Acute Chikungunya Virus Infection in Children and Adults during an Outbreak in the Maldives. Am. J. Trop. Med. Hyg. 2021, 105, 946. [Google Scholar] [CrossRef]
- Imad, H.A.; Matsee, W.; Kludkleeb, S.; Asawapaithulsert, P.; Phadungsombat, J.; Nakayama, E.E.; Suzuki, K.; Leaungwutiwong, P.; Piyaphanee, W.; Phumratanaprapin, W.; et al. Post-Chikungunya Virus Infection Musculoskeletal Disorders: Syndromic Sequelae after an Outbreak. Trop. Med. Infect. Dis. 2021, 6, 52. [Google Scholar] [CrossRef] [PubMed]
- Stecher, G.; Tamura, K.; Kumar, S. Molecular Evolutionary Genetics Analysis (MEGA) for macOS. Mol. Biol. Evol. 2020, 37, 1237–1239. [Google Scholar] [CrossRef]
- Zhang, Z.; Schwartz, S.; Wagner, L.; Miller, W. A greedy algorithm for aligning DNA sequences. J. Comput. Biol. 2000, 7, 203–214. [Google Scholar] [CrossRef]
- Trifinopoulos, J.; Nguyen, L.T.; von Haeseler, A.; Minh, B.Q. W-IQ-TREE: A fast online phylogenetic tool for maximum likelihood analysis. Nucleic Acids Res. 2016, 44, W232–W235. [Google Scholar] [CrossRef] [Green Version]
- Larsson, A. AliView: A fast and lightweight alignment viewer and editor for large datasets. Bioinformatics 2014, 30, 3276–3278. [Google Scholar] [CrossRef]
- Kosakovsky Pond, S.L.; Posada, D.; Gravenor, M.B.; Woelk, C.H.; Frost, S.D. Automated phylogenetic detection of recombination using a genetic algorithm. Mol. Biol. Evol. 2006, 23, 1891–1901. [Google Scholar] [CrossRef]
- Hapuarachchi, H.C.; Wong, W.Y.; Koo, C.; Tien, W.P.; Yeo, G.; Rajarethinam, J.; Tan, E.; Chiang, S.; Chong, C.S.; Tan, C.H.; et al. Transient transmission of Chikungunya virus in Singapore exemplifies successful mitigation of severe epidemics in a vulnerable population. Int. J. Infect. Dis. 2021, 110, 417–425. [Google Scholar] [CrossRef] [PubMed]
- Jain, J.; Kaur, N.; Haller, S.L.; Kumar, A.; Rossi, S.L.; Narayanan, V.; Kumar, D.; Gaind, R.; Weaver, S.C.; Auguste, A.J.; et al. Chikungunya Outbreaks in India: A Prospective Study Comparing Neutralization and Sequelae during Two Outbreaks in 2010 and 2016. Am. J. Trop. Med. Hyg. 2020, 102, 857–868. [Google Scholar] [CrossRef] [PubMed]
- Kariuki Njenga, M.; Nderitu, L.; Ledermann, J.P.; Ndirangu, A.; Logue, C.H.; Kelly, C.H.L.; Sang, R.; Sergon, K.; Breiman, R.; Powers, A.M. Tracking epidemic Chikungunya virus into the Indian Ocean from East Africa. J. Gen. Virol. 2008, 89, 2754–2760. [Google Scholar] [CrossRef] [PubMed]
- Kumar, N.; Mitha, M.; Krishnamoorthy, N.; Kamaraj, T.; Joseph, R.; Jambulingam, P. Genotyping of virus involved in the 2006 chikungunya outbreak in South India (Kerala and Puducherry). Curr. Sci. 2007, 93, 1412–1416. [Google Scholar]
- Khan, N.; Bhat, R.; Jain, V.; Raghavendhar, B.S.; Patel, A.K.; Nayak, K.; Chandele, A.; Murali-Krishna, K.; Ray, P. Epidemiology and molecular characterization of chikungunya virus from human cases in North India, 2016. Microbiol. Immunol. 2021, 65, 290–301. [Google Scholar] [CrossRef] [PubMed]
- Kumar, N.P.; Joseph, R.; Kamaraj, T.; Jambulingam, P. A226V mutation in virus during the 2007 chikungunya outbreak in Kerala, India. J. Gen. Virol. 2008, 89, 1945–1948. [Google Scholar] [CrossRef]
- Arankalle, V.A.; Shrivastava, S.; Cherian, S.; Gunjikar, R.S.; Walimbe, A.M.; Jadhav, S.M.; Sudeep, A.B.; Mishra, A.C. Genetic divergence of Chikungunya viruses in India (1963–2006) with special reference to the 2005–2006 explosive epidemic. J. Gen. Virol. 2007, 88, 1967–1976. [Google Scholar] [CrossRef]
- Cherian, S.S.; Walimbe, A.M.; Jadhav, S.M.; Gandhe, S.S.; Hundekar, S.L.; Mishra, A.C.; Arankalle, V.A. Evolutionary rates and timescale comparison of Chikungunya viruses inferred from the whole genome/E1 gene with special reference to the 2005-07 outbreak in the Indian subcontinent. Infect. Genet. Evol. 2009, 9, 16–23. [Google Scholar] [CrossRef]
- Sumathy, K.; Ella, K.M. Genetic diversity of Chikungunya virus, India 2006-2010: Evolutionary dynamics and serotype analyses. J. Med. Virol. 2012, 84, 462–470. [Google Scholar] [CrossRef]
- Naresh Kumar, C.V.; Sivaprasad, Y.; Sai Gopal, D.V. Genetic diversity of 2006-2009 Chikungunya virus outbreaks in Andhra Pradesh, India, reveals complete absence of E1:A226V mutation. Acta Virol. 2016, 60, 114–117. [Google Scholar] [CrossRef] [Green Version]
- Shrinet, J.; Jain, S.; Sharma, A.; Singh, S.S.; Mathur, K.; Rana, V.; Bhatnagar, R.K.; Gupta, B.; Gaind, R.; Deb, M.; et al. Genetic characterization of Chikungunya virus from New Delhi reveal emergence of a new molecular signature in Indian isolates. Virol. J. 2012, 9, 100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, A.; Kumar, A.; Uversky, V.N.; Giri, R. Understanding the interactability of chikungunya virus proteins via molecular recognition feature analysis. RSC Adv. 2018, 8, 27293–27303. [Google Scholar] [CrossRef] [Green Version]
- Voss, J.E.; Vaney, M.C.; Duquerroy, S.; Vonrhein, C.; Girard-Blanc, C.; Crublet, E.; Thompson, A.; Bricogne, G.; Rey, F.A. Glycoprotein organization of Chikungunya virus particles revealed by X-ray crystallography. Nature 2010, 468, 709–712. [Google Scholar] [CrossRef] [PubMed]
- Dutta, P.; Khan, S.; Phukan, A.; Hazarika, S.; Hazarika, N.; Chetry, S.; Khan, A.; Kaur, H. Surveillance of Chikungunya virus activity in some North-eastern states of India. Asian Pac. J. Trop. Med. 2019, 12, 19–25. [Google Scholar] [CrossRef]
- Translational Research Consortia for Chikungunya Virus in India. Current Status of Chikungunya in India. Front. Microbiol. 2021, 12, 695173. [Google Scholar] [CrossRef] [PubMed]
- Chua, C.L.; Sam, I.C.; Merits, A.; Chan, Y.F. Antigenic Variation of East/Central/South African and Asian Chikungunya Virus Genotypes in Neutralization by Immune Sera. PLoS Negl. Trop. Dis. 2016, 10, e0004960. [Google Scholar] [CrossRef] [Green Version]
- Tsetsarkin, K.A.; Chen, R.; Yun, R.; Rossi, S.L.; Plante, K.S.; Guerbois, M.; Forrester, N.; Perng, G.C.; Sreekumar, E.; Leal, G.; et al. Multi-peaked adaptive landscape for chikungunya virus evolution predicts continued fitness optimization in Aedes albopictus mosquitoes. Nat. Commun. 2014, 5, 4084. [Google Scholar] [CrossRef] [Green Version]
- Tsetsarkin, K.A.; Weaver, S.C. Sequential adaptive mutations enhance efficient vector switching by Chikungunya virus and its epidemic emergence. PLoS Pathog. 2011, 7, e1002412. [Google Scholar] [CrossRef] [Green Version]
- Anukumar, B.; Asia Devi, T.; Koshy, J.; Nikhil, N.T.; Sugunan, A.P. Molecular characterization of chikungunya virus isolates from two localized outbreaks during 2014-2019 in Kerala, India. Arch. Virol. 2021, 166, 2895–2899. [Google Scholar] [CrossRef]
- Newase, P.; More, A.; Patil, J.; Patil, P.; Jadhav, S.; Alagarasu, K.; Shah, P.; Parashar, D.; Cherian, S.S. Chikungunya phylogeography reveals persistent global transmissions of the Indian Ocean Lineage from India in association with mutational fitness. Infect. Genet. Evol. 2020, 82, 104289. [Google Scholar] [CrossRef]
- Eyase, F.; Langat, S.; Berry, I.M.; Mulwa, F.; Nyunja, A.; Mutisya, J.; Owaka, S.; Limbaso, S.; Ofula, V.; Koka, H.; et al. Emergence of a novel chikungunya virus strain bearing the E1:V80A substitution, out of the Mombasa, Kenya 2017–2018 outbreak. PLoS ONE 2020, 15, e0241754. [Google Scholar] [CrossRef]
- Ferede, G.; Tiruneh, M.; Abate, E.; Wondimeneh, Y.; Gadisa, E.; Howe, R.; Aseffa, A.; Tessema, B. Evidence of chikungunya virus infection among febrile patients in northwest Ethiopia. Int. J. Infect. Dis. 2021, 104, 183–188. [Google Scholar] [CrossRef]
- Yin, X.; Hu, T.S.; Zhang, H.; Liu, Y.; Zhou, Z.; Liu, L.; Li, P.; Wang, Y.; Yang, Z.; Yu, J.; et al. Emergent chikungunya fever and vertical transmission in Yunnan Province, China, 2019. Arch. Virol. 2021, 166, 1455–1462. [Google Scholar] [CrossRef]
- Ho, D.T.W.; Chan, D.P.C.; Lam, C.Y.; Liang, D.C.; Lee, S.S.; Kam, J.K.M. At the advancing front of Chikungunya fever in Asia: Two imported cases in Hong Kong with novel amino acid changes. J. Microbiol. Immunol. Infect. 2018, 51, 419–421. [Google Scholar] [CrossRef]
- Yang, Y.; Xu, Z.; Zheng, H.; Song, J.; Wu, Y.; Tong, Z.; Yuan, J.; Wong, G.; Liu, W.J.; Bi, Y.; et al. Genetic and Phylogenetic Characterization of a Chikungunya Virus Imported into Shenzhen, China. Virol. Sin. 2020, 35, 115–119. [Google Scholar] [CrossRef]
- Pan, J.; Fang, C.; Yan, J.; Yan, H.; Zhan, B.; Sun, Y.; Liu, Y.; Mao, H.; Cao, G.; Lv, L.; et al. Chikungunya Fever Outbreak, Zhejiang Province, China, 2017. Emerg. Infect. Dis. 2019, 25, 1589–1591. [Google Scholar] [CrossRef]
- Javelle, E.; Florescu, S.A.; Asgeirsson, H.; Jmor, S.; Eperon, G.; Leshem, E.; Blum, J.; Molina, I.; Field, V.; Pietroski, N.; et al. Increased risk of chikungunya infection in travellers to Thailand during ongoing outbreak in tourist areas: Cases imported to Europe and the Middle East, early 2019. Eurosurveillance 2019, 24, 1900146. [Google Scholar] [CrossRef] [Green Version]
- Nunes, M.R.; Faria, N.R.; de Vasconcelos, J.M.; Golding, N.; Kraemer, M.U.; de Oliveira, L.F.; Azevedo Rdo, S.; da Silva, D.E.; da Silva, E.V.; da Silva, S.P.; et al. Emergence and potential for spread of Chikungunya virus in Brazil. BMC Med. 2015, 13, 102. [Google Scholar] [CrossRef] [Green Version]
- Fortuna, C.; Toma, L.; Remoli, M.E.; Amendola, A.; Severini, F.; Boccolini, D.; Romi, R.; Venturi, G.; Rezza, G.; Di Luca, M. Vector competence of Aedes albopictus for the Indian Ocean lineage (IOL) chikungunya viruses of the 2007 and 2017 outbreaks in Italy: A comparison between strains with and without the E1:A226V mutation. Eurosurveillance 2018, 23, 1800246. [Google Scholar] [CrossRef]
- Intayot, P.; Phumee, A.; Boonserm, R.; Sor-Suwan, S.; Buathong, R.; Wacharapluesadee, S.; Brownell, N.; Poovorawan, Y.; Siriyasatien, P. Genetic Characterization of Chikungunya Virus in Field-Caught Aedes aegypti Mosquitoes Collected during the Recent Outbreaks in 2019, Thailand. Pathogens 2019, 8, 121. [Google Scholar] [CrossRef] [Green Version]
- Phumee, A.; Intayot, P.; Sor-Suwan, S.; Jittmittraphap, A.; Siriyasatien, P. Molecular detection of Indian Ocean Lineage Chikungunya virus RNA in field collected Culex quinquefasciatus Say from Bangkok, Thailand but no evidence of virus replication. PLoS ONE 2021, 16, e0246026. [Google Scholar] [CrossRef]
- Lutomiah, J.; Mulwa, F.; Mutisya, J.; Koskei, E.; Langat, S.; Nyunja, A.; Koka, H.; Konongoi, S.; Chepkorir, E.; Ofula, V.; et al. Probable contribution of Culex quinquefasciatus mosquitoes to the circulation of chikungunya virus during an outbreak in Mombasa County, Kenya, 2017–2018. Parasites Vectors 2021, 14, 138. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Strain | Collection Date | Location | Sample Type | Passage History | Accession No. |
|---|---|---|---|---|---|
| MLD19-01 | 30 March 2019 | Maldives | isolate | C6/36 | LC664141 |
| MLD19-06 | 31 March 2019 | Maldives | isolate | C6/36 | LC664142 |
| MLD19-09 | 30 March 2019 | Maldives | isolate | C6/36 | LC664143 |
| MLD19-20 | 24 April 2019 | Maldives | isolate | C6/36 | LC664144 |
| MLD19-22 | 1 April 2019 | Maldives | isolate | C6/36 | LC664145 |
| MLD19-27 | 3 April 2019 | Maldives | isolate | C6/36 | LC664146 |
| MLD19-37 | 7 April 2019 | Maldives | serum | no passage | LC664147 |
| MLD19-39 | 4 April 2019 | Maldives | isolate | C6/36 | LC664148 |
| MLD19-40 | April 2019 | Maldives | isolate | C6/36 | LC664149 |
| MLD19-47 | 10 April 2019 | Maldives | isolate | C6/36 | LC664150 |
| MLD19-53 | 14 April 2019 | Maldives | isolate | C6/36 | LC664151 |
| MLD19-68 | 19 May 2019 | Maldives | isolate | C6/36 | LC664152 |
| MLD19-70 | May 2019 | Maldives | isolate | C6/36 | LC664153 |
| MLD19-71 | May 2019 | Maldives | isolate | C6/36 | LC664154 |
| MLD19-72 | 18 June 2019 | Maldives | isolate | C6/36 | LC664155 |
| MLD19-77 | 2 August 2019 | Maldives | serum | no passage | LC664156 |
| BHTD20-WM05 | 29 June 2020 | Bangkok, Thailand | isolate | C6/36 | LC664157 |
| BHTD20-WM14 | 21 July 2020 | Bangkok, Thailand | isolate | C6/36 | LC664158 |
| BHTD20-WM19 | 4 August 2020 | Bangkok, Thailand | isolate | C6/36 | LC664159 |
| BHTD20-WM27 | 19 August 2020 | Bangkok, Thailand | serum | no passage | LC664160 |
| BHTD20-WM29 | 20 August 2020 | Bangkok, Thailand | isolate | C6/36 | LC664161 |
| BHTD20-WM34 | 26 August 2020 | Bangkok, Thailand | isolate | C6/36 | LC664162 |
| BHTD20-WM37 | 28 August 2020 | Bangkok, Thailand | serum | no passage | LC664163 |
| BHTD20-WM40 | 8 September 2020 | Bangkok, Thailand | serum | no passage | LC664164 |
| BHTD20-WM44 | 13 September 2020 | Bangkok, Thailand | serum | no passage | LC664165 |
| BHTD20-WM48 | 25 September 2020 | Bangkok, Thailand | serum | no passage | LC664166 |
| Year | Local Strain | Travel-Associated Strain | ||
|---|---|---|---|---|
| Region | Location | Region | Reported Country (Origin Country) | |
| 2009 | Southeast Asia | Singapore * 1 (1) | ||
| 2010 | Indian subcontinent | India 20 (15) | Europe | France (India) 1 (0) |
| Europe | France 1 (0) | East Asia | China (India) 4 (4) | |
| Southeast Asia | Singapore * 4 (4) | |||
| 2011 | Indian subcontinent | India 8 (5) | ||
| Southeast Asia | Singapore * 2 (2) | |||
| 2012 | Indian subcontinent | India 16 (10) | ||
| Southeast Asia | Singapore * 3 (3) | |||
| 2013 | Indian subcontinent | India 6 (6) | ||
| Southeast Asia | Singapore * 2 (2) | |||
| 2014 | Indian subcontinent | India 4 (4) | ||
| Africa | Kenya 2 (2) | |||
| 2015 | Indian subcontinent | India 5 (5) | ||
| Southeast Asia | Singapore * 1 (1) | |||
| Africa | Kenya 1 (1) | |||
| 2016 | Indian subcontinent | India 27 (27), Pakistan 8 (7) | Pacific | Australia (India) 1 (1) |
| Africa | Kenya 15 (12) | East Asia | Hong Kong (India) 2 (2) | |
| 2017 | Indian subcontinent | India 4 (4), Pakistan 5 (1), Bangladesh 37 (37) | East Asia | China (Pakistan) 1 (1), (Bangladesh) 2 (2) |
| Africa | Kenya 5 (5) | Pacific | Australia (Bangladesh) 1 (1) | |
| Europe | Italy 10 (10) | |||
| Southeast Asia | Singapore * 1 (1) | |||
| 2018 | Indian subcontinent | India 2 (2) | Europe | Slovenia (Thailand) 1 (1) |
| Africa | Kenya 19 (19), Sudan 80 (80) | |||
| Southeast Asia | Thailand 15 (15) | |||
| 2019 | Indian subcontinent | India 1 (1), Maldives 16 (16) | Pacific | Australia (Thailand) 1 (1) |
| Africa | Djibouti 1 (1) | East Asia | China (Myanmar 10 (10), Thailand 1 (1)) Taiwan (Myanmar 7 (0), Thailand 4 (0), Malaysia 1 (0)) | |
| Southeast Asia | Thailand 29 (29) | Europe | Finland (Thailand) 2 (2) | |
| East Asia | China 7 (7), Taiwan 8 (0) | |||
| 2020 | Southeast Asia | Thailand 21 (21) | ||
| Codon Site | MEME p < 0.1 | FUBAR pp > 0.9 | FEL p < 0.1 | SLAC p < 0.1 | Amino Acid Substitution | Sequences with Derived Amino Acid State |
|---|---|---|---|---|---|---|
| Nonstructural proteins | ||||||
| 171 | 0.00 | 0.999 | 0.002 | 0.006 | nsP1-R171Q | Comoros 2005: HQ456252 Italy 2007: MK120202, KX262993 Sri Lanka 2006: AB455493 India 2016: MW321606, MG137428; 2017: MK286893 Kenya 2016: MH423803; 2017: MT380161 |
| 665 | 0.12 | 0.946 | 0.095 | 0.131 | nsP2-H130Y | Singapore 2009: MH647212; 2010: MH647214 2012: MH647182, MH647184; 2013: MH647187 India 2014: MW042254; 2015: MK370031 IE clade virus *, IS clade virus * |
| Structural proteins | ||||||
| 24 | 0.09 | 0.893 | 0.069 | 0.296 | C-T24A | Laos 2013: MF076569 Djibouti 2019: MT023791 Thailand 2019: LC580269; 2020: BHTD20-WM14, BHTD20-WM19 |
| 471 | 0.03 | 0.995 | 0.022 | 0.079 | E2-Q146R | Seychelles 2005: AM258991 Bangladesh 2017: MG912993 Kenya 2018: MT526797 Thailand 2019: MN075150; 2020: BHTD20-WM05 |
| 546 | 0.12 | 0.965 | 0.097 | 0.237 | E2-K221R | India 2012: MW581867 Bangladesh 2017: LC580241, LC580244 Thailand 2019: MN075150; 2020: BHTD20-WM27 |
| 795 | 0.07 | 0.902 | 0.061 | 0.296 | 6K-A47V 6K-A47G | India 2016: MK473625 Pakistan 2016: MF774613 Singapore 2013: MH647192; 2014: MH647202 |
| 813 | 0.12 | 0.946 | 0.093 | 0.198 | E1-V4A | India 2007: EU372006; 2008: GQ428215; 2009: KT336777 2015: KX619425, KX619424 2016: MK518340, MK551552, MK551553, MK473628 Bangladesh 2008: FJ807898; 2011: KU365371 Singapore 2013: MH647192; 2014: MH647202 Thailand 2020: BHTD20-WM44, BHTD20-WM27, BHTD20-WM48 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Phadungsombat, J.; Imad, H.A.; Nakayama, E.E.; Leaungwutiwong, P.; Ramasoota, P.; Nguitragool, W.; Matsee, W.; Piyaphanee, W.; Shioda, T. Spread of a Novel Indian Ocean Lineage Carrying E1-K211E/E2-V264A of Chikungunya Virus East/Central/South African Genotype across the Indian Subcontinent, Southeast Asia, and Eastern Africa. Microorganisms 2022, 10, 354. https://doi.org/10.3390/microorganisms10020354
Phadungsombat J, Imad HA, Nakayama EE, Leaungwutiwong P, Ramasoota P, Nguitragool W, Matsee W, Piyaphanee W, Shioda T. Spread of a Novel Indian Ocean Lineage Carrying E1-K211E/E2-V264A of Chikungunya Virus East/Central/South African Genotype across the Indian Subcontinent, Southeast Asia, and Eastern Africa. Microorganisms. 2022; 10(2):354. https://doi.org/10.3390/microorganisms10020354
Chicago/Turabian StylePhadungsombat, Juthamas, Hisham A. Imad, Emi E. Nakayama, Pornsawan Leaungwutiwong, Pongrama Ramasoota, Wang Nguitragool, Wasin Matsee, Watcharapong Piyaphanee, and Tatsuo Shioda. 2022. "Spread of a Novel Indian Ocean Lineage Carrying E1-K211E/E2-V264A of Chikungunya Virus East/Central/South African Genotype across the Indian Subcontinent, Southeast Asia, and Eastern Africa" Microorganisms 10, no. 2: 354. https://doi.org/10.3390/microorganisms10020354