

Group B Streptococcus: Virulence Factors and Pathogenic Mechanism
Abstract
:1. Introduction
2. Virulence Factors Associated with Interaction with the Vagina
2.1. Adhesins
2.2. Hemolytic Pigment
3. Virulence Factors Associated with Interaction with the Cervix
3.1. Alpha C Protein
3.2. Capsule
4. Virulence Factors Associated with Interaction with the Endometrium
5. Virulence Factors Associated with Interaction with the Feto–Maternal Interface
Membrane Vesicles (MVs)
6. Effects on the Newborn
6.1. HvgA
6.2. CAMP Factor
7. GBS Vaccine
8. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Russell, N.J.; Seale, A.C.; O’Driscoll, M.; O’Sullivan, C.; Bianchi-Jassir, F.; Gonzalez-Guarin, J.; Lawn, J.E.; Baker, C.J.; Bartlett, L.; Cutland, C.; et al. Maternal Colonization with Group B Streptococcus and Serotype Distribution Worldwide: Systematic Review and Meta-analyses. Clin. Infect. Dis. 2017, 65 (Suppl. S2), S100–S111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madrid, L.; Seale, A.C.; Kohli-Lynch, M.; Edmond, K.M.; Lawn, J.E.; Heath, P.T.; Madhi, S.A.; Baker, C.J.; Bartlett, L.; Cutland, C.; et al. Infant Group B Streptococcal Disease Incidence and Serotypes Worldwide: Systematic Review and Meta-analyses. Clin. Infect. Dis. 2017, 65 (Suppl. S2), S160–S172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raabe, V.N.; Shane, A.L. Group B Streptococcus (Streptococcus agalactiae). Microbiol. Spectr. 2019, 7, GPP3-0007-2018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lawn, J.E.; Bianchi-Jassir, F.; Russell, N.J.; Kohli-Lynch, M.; Tann, C.J.; Hall, J.; Madrid, L.; Baker, C.J.; Bartlett, L.; Cutland, C.; et al. Group B Streptococcal Disease Worldwide for Pregnant Women, Stillbirths, and Children: Why, What, and How to Undertake Estimates? Clin. Infect. Dis. 2017, 65 (Suppl. S2), S89–S99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verani, J.R.; McGee, L.; Schrag, S.J. Prevention of perinatal group B streptococcal disease--revised guidelines from CDC, 2010. MMWR Recomm. Rep. 2010, 59, 1–36. [Google Scholar]
- Dhudasia, M.B.; Flannery, D.D.; Pfeifer, M.R.; Puopolo, K.M. Updated Guidance: Prevention and Management of Perinatal Group B Streptococcus Infection. Neoreviews 2021, 22, e177–e188. [Google Scholar] [CrossRef]
- Prevention of perinatal group B streptococcal disease: A public health perspective. Centers for Disease Control and Prevention. MMWR Recomm. Rep. 1996, 45, 1–24. [Google Scholar]
- Revised guidelines for prevention of early-onset group B streptococcal (GBS) infection. American Academy of Pediatrics Committee on Infectious Diseases and Committee on Fetus and Newborn. Pediatrics 1997, 99, 489–496. [Google Scholar]
- Centers for Disease Control and Prevention. Perinatal group B streptococcal disease after universal screening recommendations–United States, 2003–2005. MMWR Morb. Mortal Wkly. Rep. 2007, 56, 701–705. [Google Scholar]
- Blencowe, H.; Cousens, S.; Jassir, F.B.; Say, L.; Chou, D.; Mathers, C.; Hogan, D.; Shiekh, S.; Qureshi, Z.U.; You, D.; et al. National, regional, and worldwide estimates of stillbirth rates in 2015, with trends from 2000: A systematic analysis. Lancet Glob. Health 2016, 4, e98–e108. [Google Scholar] [CrossRef] [Green Version]
- Lawn, J.E.; Blencowe, H.; Oza, S.; You, D.; Lee, A.C.; Waiswa, P.; Lalli, M.; Bhutta, Z.; Barros, A.J.; Christian, P.; et al. Every Newborn: Progress, priorities, and potential beyond survival. Lancet 2014, 384, 189–205. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Oza, S.; Hogan, D.; Perin, J.; Rudan, I.; Lawn, J.E.; Cousens, S.; Mathers, C.; Black, R.E. Global, regional, and national causes of child mortality in 2000–2013, with projections to inform post-2015 priorities: An updated systematic analysis. Lancet 2015, 385, 430–440. [Google Scholar] [CrossRef] [PubMed]
- Puopolo, K.M.; Lynfield, R.; Cummings, J.J.; Committee on Fetus and Newborn. Management of Infants at Risk for Group B Streptococcal Disease. Pediatrics 2019, 144, e20191881. [Google Scholar] [CrossRef] [Green Version]
- Di Renzo, G.C.; Melin, P.; Berardi, A.; Blennow, M.; Carbonell-Estrany, X.; Donzelli, G.P.; Hakansson, S.; Hod, M.; Hughes, R.; Kurtzer, M.; et al. Intrapartum GBS screening and antibiotic prophylaxis: A European consensus conference. J. Matern. Fetal Neonatal Med. 2015, 28, 766–782. [Google Scholar] [CrossRef] [PubMed]
- Seo, H.S.; Minasov, G.; Seepersaud, R.; Doran, K.S.; Dubrovska, I.; Shuvalova, L.; Anderson, W.F.; Iverson, T.M.; Sullam, P.M. Characterization of fibrinogen binding by glycoproteins Srr1 and Srr2 of Streptococcus agalactiae. J. Biol. Chem. 2013, 288, 35982–35996. [Google Scholar] [CrossRef] [Green Version]
- Pietrocola, G.; Arciola, C.R.; Rindi, S.; Montanaro, L.; Speziale, P. Streptococcus agalactiae Non-Pilus, Cell Wall-Anchored Proteins: Involvement in Colonization and Pathogenesis and Potential as Vaccine Candidates. Front. Immunol. 2018, 9, 602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tazi, A.; Disson, O.; Bellais, S.; Bouaboud, A.; Dmytruk, N.; Dramsi, S.; Mistou, M.Y.; Khun, H.; Mechler, C.; Tardieux, I.; et al. The surface protein HvgA mediates group B streptococcus hypervirulence and meningeal tropism in neonates. J. Exp. Med. 2010, 207, 2313–2322. [Google Scholar] [CrossRef]
- Whidbey, C.; Harrell, M.I.; Burnside, K.; Ngo, L.; Becraft, A.K.; Iyer, L.M.; Aravind, L.; Hitti, J.; Adams Waldorf, K.M.; Rajagopal, L. A hemolytic pigment of Group B Streptococcus allows bacterial penetration of human placenta. J. Exp. Med. 2013, 210, 1265–1281. [Google Scholar] [CrossRef] [Green Version]
- Whidbey, C.; Vornhagen, J.; Gendrin, C.; Boldenow, E.; Samson, J.M.; Doering, K.; Ngo, L.; Ezekwe, E.A., Jr.; Gundlach, J.H.; Elovitz, M.A.; et al. A streptococcal lipid toxin induces membrane permeabilization and pyroptosis leading to fetal injury. EMBO Mol. Med. 2015, 7, 488–505. [Google Scholar] [CrossRef] [Green Version]
- Nanduri, S.A.; Petit, S.; Smelser, C.; Apostol, M.; Alden, N.B.; Harrison, L.H.; Lynfield, R.; Vagnone, P.S.; Burzlaff, K.; Spina, N.L.; et al. Epidemiology of Invasive Early-Onset and Late-Onset Group B Streptococcal Disease in the United States, 2006 to 2015: Multistate Laboratory and Population-Based Surveillance. JAMA Pediatr. 2019, 173, 224–233. [Google Scholar] [CrossRef]
- Armistead, B.; Oler, E.; Adams Waldorf, K.; Rajagopal, L. The Double Life of Group B Streptococcus: Asymptomatic Colonizer and Potent Pathogen. J. Mol. Biol. 2019, 431, 2914–2931. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.M.; Gori, A.; Nobbs, A.H.; Heyderman, R.S. Streptococcal Serine-Rich Repeat Proteins in Colonization and Disease. Front. Microbiol. 2020, 11, 593356. [Google Scholar] [CrossRef] [PubMed]
- Seepersaud, R.; Anderson, A.C.; Bensing, B.A.; Choudhury, B.P.; Clarke, A.J.; Sullam, P.M. O-acetylation controls the glycosylation of bacterial serine-rich repeat glycoproteins. J. Biol. Chem. 2021, 296, 100249. [Google Scholar] [CrossRef]
- Mistou, M.Y.; Dramsi, S.; Brega, S.; Poyart, C.; Trieu-Cuot, P. Molecular dissection of the secA2 locus of group B Streptococcus reveals that glycosylation of the Srr1 LPXTG protein is required for full virulence. J. Bacteriol. 2009, 191, 4195–4206. [Google Scholar] [CrossRef] [PubMed]
- Six, A.; Bellais, S.; Bouaboud, A.; Fouet, A.; Gabriel, C.; Tazi, A.; Dramsi, S. Srr2, a multifaceted adhesin expressed by ST-17 hypervirulent Group B Streptococcus involved in binding to both fibrinogen and plasminogen. Mol. Microbiol. 2015, 97, 1209–1222. [Google Scholar] [CrossRef] [PubMed]
- Seifert, K.N.; Adderson, E.E.; Whiting, A.A.; Bohnsack, J.F.; Crowley, P.J.; Brady, L.J. A unique serine-rich repeat protein (Srr-2) and novel surface antigen (epsilon) associated with a virulent lineage of serotype III Streptococcus agalactiae. Microbiology 2006, 152 Pt 4, 1029–1040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gori, A.; Harrison, O.B.; Mlia, E.; Nishihara, Y.; Chan, J.M.; Msefula, J.; Mallewa, M.; Dube, Q.; Swarthout, T.D.; Nobbs, A.H.; et al. Pan-GWAS of Streptococcus agalactiae Highlights Lineage-Specific Genes Associated with Virulence and Niche Adaptation. mBio 2020, 11, e00728-20. [Google Scholar] [CrossRef] [PubMed]
- Sheen, T.R.; Jimenez, A.; Wang, N.Y.; Banerjee, A.; van Sorge, N.M.; Doran, K.S. Serine-rich repeat proteins and pili promote Streptococcus agalactiae colonization of the vaginal tract. J. Bacteriol. 2011, 193, 6834–6842. [Google Scholar] [CrossRef] [Green Version]
- Lacasse, M.; Valentin, A.S.; Corvec, S.; Bemer, P.; Jolivet-Gougeon, A.; Plouzeau, C.; Tande, D.; Mereghetti, L.; Bernard, L.; Lartigue, M.F.; et al. Genotypic Characterization and Biofilm Production of Group B Streptococcus Strains Isolated from Bone and Joint Infections. Microbiol. Spectr. 2022, 10, e0232921. [Google Scholar] [CrossRef]
- Wang, N.Y.; Patras, K.A.; Seo, H.S.; Cavaco, C.K.; Rosler, B.; Neely, M.N.; Sullam, P.M.; Doran, K.S. Group B streptococcal serine-rich repeat proteins promote interaction with fibrinogen and vaginal colonization. J. Infect. Dis. 2014, 210, 982–991. [Google Scholar] [CrossRef] [Green Version]
- Seale, A.C.; Bianchi-Jassir, F.; Russell, N.J.; Kohli-Lynch, M.; Tann, C.J.; Hall, J.; Madrid, L.; Blencowe, H.; Cousens, S.; Baker, C.J.; et al. Estimates of the Burden of Group B Streptococcal Disease Worldwide for Pregnant Women, Stillbirths, and Children. Clin. Infect. Dis. 2017, 65 (Suppl. S2), S200–S219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beckert, S.; Kreikemeyer, B.; Podbielski, A. Group A streptococcal rofA gene is involved in the control of several virulence genes and eukaryotic cell attachment and internalization. Infect. Immun. 2001, 69, 534–537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samen, U.; Heinz, B.; Boisvert, H.; Eikmanns, B.J.; Reinscheid, D.J.; Borges, F. Rga is a regulator of adherence and pilus formation in Streptococcus agalactiae. Microbiology 2011, 157 Pt 8, 2319–2327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buscetta, M.; Papasergi, S.; Firon, A.; Pietrocola, G.; Biondo, C.; Mancuso, G.; Midiri, A.; Romeo, L.; Teti, G.; Speziale, P.; et al. FbsC, a novel fibrinogen-binding protein, promotes Streptococcus agalactiae-host cell interactions. J. Biol. Chem. 2014, 289, 21003–21015. [Google Scholar] [CrossRef]
- Bobadilla, F.J.; Novosak, M.G.; Cortese, I.J.; Delgado, O.D.; Laczeski, M.E. Prevalence, serotypes and virulence genes of Streptococcus agalactiae isolated from pregnant women with 35-37 weeks of gestation. BMC Infect. Dis. 2021, 21, 73. [Google Scholar] [CrossRef]
- Gutekunst, H.; Eikmanns, B.J.; Reinscheid, D.J. The novel fibrinogen-binding protein FbsB promotes Streptococcus agalactiae invasion into epithelial cells. Infect. Immun. 2004, 72, 3495–3504. [Google Scholar] [CrossRef] [Green Version]
- Buscetta, M.; Firon, A.; Pietrocola, G.; Biondo, C.; Mancuso, G.; Midiri, A.; Romeo, L.; Galbo, R.; Venza, M.; Venza, I.; et al. PbsP, a cell wall-anchored protein that binds plasminogen to promote hematogenous dissemination of group B Streptococcus. Mol. Microbiol. 2016, 101, 27–41. [Google Scholar] [CrossRef] [Green Version]
- Cook, L.C.C.; Hu, H.; Maienschein-Cline, M.; Federle, M.J. A Vaginal Tract Signal Detected by the Group B Streptococcus SaeRS System Elicits Transcriptomic Changes and Enhances Murine Colonization. Infect. Immun. 2018, 86, e00762-17. [Google Scholar] [CrossRef] [Green Version]
- Coppolino, F.; Romeo, L.; Pietrocola, G.; Lentini, G.; De Gaetano, G.V.; Teti, G.; Galbo, R.; Beninati, C. Lysine Residues in the MK-Rich Region Are Not Required for Binding of the PbsP Protein From Group B Streptococci to Plasminogen. Front. Cell Infect. Microbiol. 2021, 11, 679792. [Google Scholar] [CrossRef]
- De Gaetano, G.V.; Pietrocola, G.; Romeo, L.; Galbo, R.; Lentini, G.; Giardina, M.; Biondo, C.; Midiri, A.; Mancuso, G.; Venza, M.; et al. The Streptococcus agalactiae cell wall-anchored protein PbsP mediates adhesion to and invasion of epithelial cells by exploiting the host vitronectin/alphav integrin axis. Mol. Microbiol. 2018, 110, 82–94. [Google Scholar] [CrossRef]
- Dramsi, S.; Caliot, E.; Bonne, I.; Guadagnini, S.; Prevost, M.C.; Kojadinovic, M.; Lalioui, L.; Poyart, C.; Trieu-Cuot, P. Assembly and role of pili in group B streptococci. Mol. Microbiol. 2006, 60, 1401–1413. [Google Scholar] [CrossRef] [PubMed]
- Konto-Ghiorghi, Y.; Mairey, E.; Mallet, A.; Dumenil, G.; Caliot, E.; Trieu-Cuot, P.; Dramsi, S. Dual role for pilus in adherence to epithelial cells and biofilm formation in Streptococcus agalactiae. PLoS Pathog. 2009, 5, e1000422. [Google Scholar] [CrossRef] [PubMed]
- Nobbs, A.H.; Rosini, R.; Rinaudo, C.D.; Maione, D.; Grandi, G.; Telford, J.L. Sortase A utilizes an ancillary protein anchor for efficient cell wall anchoring of pili in Streptococcus agalactiae. Infect. Immun. 2008, 76, 3550–3560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lauer, P.; Rinaudo, C.D.; Soriani, M.; Margarit, I.; Maione, D.; Rosini, R.; Taddei, A.R.; Mora, M.; Rappuoli, R.; Grandi, G.; et al. Genome analysis reveals pili in Group B Streptococcus. Science 2005, 309, 105. [Google Scholar] [CrossRef]
- Rosini, R.; Rinaudo, C.D.; Soriani, M.; Lauer, P.; Mora, M.; Maione, D.; Taddei, A.; Santi, I.; Ghezzo, C.; Brettoni, C.; et al. Identification of novel genomic islands coding for antigenic pilus-like structures in Streptococcus agalactiae. Mol. Microbiol. 2006, 61, 126–141. [Google Scholar] [CrossRef] [PubMed]
- Nabavinia, M.; Khalili, M.B.; Sadeh, M.; Eslami, G.; Vakili, M.; Azartoos, N.; Mojibiyan, M. Distribution of Pilus island and antibiotic resistance genes in Streptococcus agalactiae obtained from vagina of pregnant women in Yazd, Iran. Iran J. Microbiol. 2020, 12, 411–416. [Google Scholar] [CrossRef]
- Spellerberg, B.; Rozdzinski, E.; Martin, S.; Weber-Heynemann, J.; Schnitzler, N.; Lutticken, R.; Podbielski, A. Lmb, a protein with similarities to the LraI adhesin family, mediates attachment of Streptococcus agalactiae to human laminin. Infect. Immun. 1999, 67, 871–878. [Google Scholar] [CrossRef] [Green Version]
- Zygiel, E.M.; Nolan, E.M. Transition Metal Sequestration by the Host-Defense Protein Calprotectin. Annu. Rev. Biochem. 2018, 87, 621–643. [Google Scholar] [CrossRef]
- Burcham, L.R.; Le Breton, Y.; Radin, J.N.; Spencer, B.L.; Deng, L.; Hiron, A.; Ransom, M.R.; Mendonca, J.D.C.; Belew, A.T.; El-Sayed, N.M.; et al. Identification of Zinc-Dependent Mechanisms Used by Group B Streptococcus To Overcome Calprotectin-Mediated Stress. mBio 2020, 11, e02302-20. [Google Scholar] [CrossRef]
- Cheng, Q.; Stafslien, D.; Purushothaman, S.S.; Cleary, P. The group B streptococcal C5a peptidase is both a specific protease and an invasin. Infect. Immun. 2002, 70, 2408–2413. [Google Scholar] [CrossRef] [Green Version]
- Chmouryguina, I.; Suvorov, A.; Ferrieri, P.; Cleary, P.P. Conservation of the C5a peptidase genes in group A and B streptococci. Infect. Immun. 1996, 64, 2387–2390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khodaei, F.; Najafi, M.; Hasani, A.; Kalantar, E.; Sharifi, E.; Amini, A.; Aghazadeh, M. Pilus-encoding islets in S. agalactiae and its association with antibacterial resistance and serotype distribution. Microb. Pathog. 2018, 116, 189–194. [Google Scholar] [CrossRef] [PubMed]
- Lopez, Y.; Parra, E.; Cepas, V.; Sanfeliu, I.; Juncosa, T.; Andreu, A.; Xercavins, M.; Perez, J.; Sanz, S.; Vergara, A.; et al. Serotype, virulence profile, antimicrobial resistance and macrolide-resistance determinants in Streptococcus agalactiae isolates in pregnant women and neonates in Catalonia, Spain. Enferm. Infecc. Microbiol. Clin. (Engl. Ed.) 2018, 36, 472–477. [Google Scholar] [CrossRef] [PubMed]
- Piliponsky, A.M.; Sharma, K.; Quach, P.; Brokaw, A.; Nguyen, S.; Orvis, A.; Saha, S.S.; Samanas, N.B.; Seepersaud, R.; Tang, Y.P.; et al. Mast cell derived factor XIIIA contributes to sexual dimorphic defense against group B Streptococcal infections. J. Clin. Investig. 2022, 132, e157999. [Google Scholar] [CrossRef]
- Vornhagen, J.; Quach, P.; Boldenow, E.; Merillat, S.; Whidbey, C.; Ngo, L.Y.; Adams Waldorf, K.M.; Rajagopal, L. Bacterial Hyaluronidase Promotes Ascending GBS Infection and Preterm Birth. mBio 2016, 7, e00781-16. [Google Scholar] [CrossRef] [Green Version]
- Kurian, N.K.; Modi, D. Mechanisms of group B Streptococcus-mediated preterm birth: Lessons learnt from animal models. Reprod. Fertil. 2022, 3, R109–R120. [Google Scholar] [CrossRef]
- Gendrin, C.; Vornhagen, J.; Armistead, B.; Singh, P.; Whidbey, C.; Merillat, S.; Knupp, D.; Parker, R.; Rogers, L.M.; Quach, P.; et al. A Nonhemolytic Group B Streptococcus Strain Exhibits Hypervirulence. J. Infect. Dis. 2018, 217, 983–987. [Google Scholar] [CrossRef]
- Coleman, M.; Armistead, B.; Orvis, A.; Quach, P.; Brokaw, A.; Gendrin, C.; Sharma, K.; Ogle, J.; Merillat, S.; Dacanay, M.; et al. Hyaluronidase Impairs Neutrophil Function and Promotes Group B Streptococcus Invasion and Preterm Labor in Nonhuman Primates. mBio 2021, 12, e03115-20. [Google Scholar] [CrossRef]
- Rosa-Fraile, M.; Rodriguez-Granger, J.; Haidour-Benamin, A.; Cuerva, J.M.; Sampedro, A. Granadaene: Proposed structure of the group B Streptococcus polyenic pigment. Appl. Environ. Microbiol. 2006, 72, 6367–6370. [Google Scholar] [CrossRef] [Green Version]
- Pritzlaff, C.A.; Chang, J.C.; Kuo, S.P.; Tamura, G.S.; Rubens, C.E.; Nizet, V. Genetic basis for the beta-haemolytic/cytolytic activity of group B Streptococcus. Mol. Microbiol. 2001, 39, 236–247. [Google Scholar] [CrossRef]
- Spellerberg, B.; Pohl, B.; Haase, G.; Martin, S.; Weber-Heynemann, J.; Lutticken, R. Identification of genetic determinants for the hemolytic activity of Streptococcus agalactiae by ISS1 transposition. J. Bacteriol. 1999, 181, 3212–3219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armistead, B.; Whidbey, C.; Iyer, L.M.; Herrero-Foncubierta, P.; Quach, P.; Haidour, A.; Aravind, L.; Cuerva, J.M.; Jaspan, H.B.; Rajagopal, L. The cyl Genes Reveal the Biosynthetic and Evolutionary Origins of the Group B Streptococcus Hemolytic Lipid, Granadaene. Front. Microbiol. 2019, 10, 3123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boldenow, E.; Gendrin, C.; Ngo, L.; Bierle, C.; Vornhagen, J.; Coleman, M.; Merillat, S.; Armistead, B.; Whidbey, C.; Alishetti, V.; et al. Group B Streptococcus circumvents neutrophils and neutrophil extracellular traps during amniotic cavity invasion and preterm labor. Sci. Immunol. 2016, 1, eaah4576. [Google Scholar] [CrossRef] [PubMed]
- Jusuf, S.; Dong, P.T.; Hui, J.; Ulloa, E.R.; Liu, G.Y.; Cheng, J.X. Granadaene Photobleaching Reduces the Virulence and Increases Antimicrobial Susceptibility of Streptococcus agalactiae. Photochem. Photobiol. 2021, 97, 816–825. [Google Scholar] [CrossRef]
- Jahn, K.; Shumba, P.; Quach, P.; Musken, M.; Wesche, J.; Greinacher, A.; Rajagopal, L.; Hammerschmidt, S.; Siemens, N. Group B Streptococcal Hemolytic Pigment Impairs Platelet Function in a Two-Step Process. Cells 2022, 11, 1637. [Google Scholar] [CrossRef]
- Baron, M.J.; Filman, D.J.; Prophete, G.A.; Hogle, J.M.; Madoff, L.C. Identification of a glycosaminoglycan binding region of the alpha C protein that mediates entry of group B Streptococci into host cells. J. Biol. Chem. 2007, 282, 10526–10536. [Google Scholar] [CrossRef] [Green Version]
- Puopolo, K.M.; Hollingshead, S.K.; Carey, V.J.; Madoff, L.C. Tandem repeat deletion in the alpha C protein of group B streptococcus is recA independent. Infect. Immun. 2001, 69, 5037–5045. [Google Scholar] [CrossRef] [Green Version]
- Noble, K.; Lu, J.; Guevara, M.A.; Doster, R.S.; Chambers, S.A.; Rogers, L.M.; Moore, R.E.; Spicer, S.K.; Eastman, A.J.; Francis, J.D.; et al. Group B Streptococcus cpsE Is Required for Serotype V Capsule Production and Aids in Biofilm Formation and Ascending Infection of the Reproductive Tract during Pregnancy. ACS Infect. Dis. 2021, 7, 2686–2696. [Google Scholar] [CrossRef]
- Rostami, S.; Moeineddini, L.; Ghandehari, F.; Khorasani, M.R.; Shoaei, P.; Ebrahimi, N. Macrolide-resistance, capsular genotyping and associated factors of group B Streptococci colonized pregnant women in Isfahan, Iran. Iran J. Microbiol. 2021, 13, 183–189. [Google Scholar] [CrossRef]
- Abeyta, M.; Hardy, G.G.; Yother, J. Genetic alteration of capsule type but not PspA type affects accessibility of surface-bound complement and surface antigens of Streptococcus pneumoniae. Infect. Immun. 2003, 71, 218–225. [Google Scholar] [CrossRef] [Green Version]
- Winkelstein, J.A.; Abramovitz, A.S.; Tomasz, A. Activation of C3 via the alternative complement pathway results in fixation of C3b to the pneumococcal cell wall. J. Immunol. 1980, 124, 2502–2506. [Google Scholar] [PubMed]
- Wessels, M.R.; Rubens, C.E.; Benedi, V.J.; Kasper, D.L. Definition of a bacterial virulence factor: Sialylation of the group B streptococcal capsule. Proc. Natl. Acad. Sci. USA 1989, 86, 8983–8987. [Google Scholar] [CrossRef] [Green Version]
- Tang, Y.Q.; Yeaman, M.R.; Selsted, M.E. Antimicrobial peptides from human platelets. Infect. Immun. 2002, 70, 6524–6533. [Google Scholar] [CrossRef] [Green Version]
- Carlin, A.F.; Chang, Y.C.; Areschoug, T.; Lindahl, G.; Hurtado-Ziola, N.; King, C.C.; Varki, A.; Nizet, V. Group B Streptococcus suppression of phagocyte functions by protein-mediated engagement of human Siglec-5. J. Exp. Med. 2009, 206, 1691–1699. [Google Scholar] [CrossRef] [PubMed]
- Uchiyama, S.; Sun, J.; Fukahori, K.; Ando, N.; Wu, M.; Schwarz, F.; Siddiqui, S.S.; Varki, A.; Marth, J.D.; Nizet, V. Dual actions of group B Streptococcus capsular sialic acid provide resistance to platelet-mediated antimicrobial killing. Proc. Natl. Acad. Sci. USA 2019, 116, 7465–7470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carlin, A.F.; Uchiyama, S.; Chang, Y.C.; Lewis, A.L.; Nizet, V.; Varki, A. Molecular mimicry of host sialylated glycans allows a bacterial pathogen to engage neutrophil Siglec-9 and dampen the innate immune response. Blood 2009, 113, 3333–3336. [Google Scholar] [CrossRef]
- Carlin, A.F.; Lewis, A.L.; Varki, A.; Nizet, V. Group B streptococcal capsular sialic acids interact with siglecs (immunoglobulin-like lectins) on human leukocytes. J. Bacteriol. 2007, 189, 1231–1237. [Google Scholar] [CrossRef] [Green Version]
- Secundino, I.; Lizcano, A.; Roupe, K.M.; Wang, X.; Cole, J.N.; Olson, J.; Ali, S.R.; Dahesh, S.; Amayreh, L.K.; Henningham, A.; et al. Host and pathogen hyaluronan signal through human siglec-9 to suppress neutrophil activation. J. Mol. Med. 2016, 94, 219–233. [Google Scholar] [CrossRef] [Green Version]
- Critchley, H.O.D.; Maybin, J.A.; Armstrong, G.M.; Williams, A.R.W. Physiology of the Endometrium and Regulation of Menstruation. Physiol. Rev. 2020, 100, 1149–1179. [Google Scholar] [CrossRef]
- Eastman, A.J.; Vrana, E.N.; Grimaldo, M.T.; Jones, A.D.; Rogers, L.M.; Alcendor, D.J.; Aronoff, D.M. Cytotrophoblasts suppress macrophage-mediated inflammation through a contact-dependent mechanism. Am. J. Reprod. Immunol. 2021, 85, e13352. [Google Scholar] [CrossRef]
- McCutcheon, C.R.; Pell, M.E.; Gaddy, J.A.; Aronoff, D.M.; Petroff, M.G.; Manning, S.D. Production and Composition of Group B Streptococcal Membrane Vesicles Vary Across Diverse Lineages. Front. Microbiol. 2021, 12, 770499. [Google Scholar] [CrossRef] [PubMed]
- Surve, M.V.; Anil, A.; Kamath, K.G.; Bhutda, S.; Sthanam, L.K.; Pradhan, A.; Srivastava, R.; Basu, B.; Dutta, S.; Sen, S.; et al. Membrane Vesicles of Group B Streptococcus Disrupt Feto-Maternal Barrier Leading to Preterm Birth. PLoS Pathog. 2016, 12, e1005816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armistead, B.; Quach, P.; Snyder, J.M.; Santana-Ufret, V.; Furuta, A.; Brokaw, A.; Rajagopal, L. Hemolytic Membrane Vesicles of Group B Streptococcus Promote Infection. J. Infect. Dis. 2021, 223, 1488–1496. [Google Scholar] [CrossRef] [PubMed]
- Elling, R.; Hufnagel, M.; de Zoysa, A.; Lander, F.; Zumstein, K.; Krueger, M.; Henneke, P. Synchronous recurrence of group B streptococcal late-onset sepsis in twins. Pediatrics 2014, 133, e1388-91. [Google Scholar] [CrossRef]
- Kim, H.J.; Kim, S.Y.; Seo, W.H.; Choi, B.M.; Yoo, Y.; Lee, K.H.; Eun, B.L.; Kim, H.J. Outbreak of late-onset group B streptococcal infections in healthy newborn infants after discharge from a maternity hospital: A case report. J. Korean Med. Sci. 2006, 21, 347–350. [Google Scholar] [CrossRef] [Green Version]
- Bekker, V.; Bijlsma, M.W.; van de Beek, D.; Kuijpers, T.W.; van der Ende, A. Incidence of invasive group B streptococcal disease and pathogen genotype distribution in newborn babies in the Netherlands over 25 years: A nationwide surveillance study. Lancet Infect. Dis. 2014, 14, 1083–1089. [Google Scholar] [CrossRef]
- Awwad, E.; Srour, M.; Hasan, S.; Khatib, S. Molecular determination, serotyping, antibiotic profile and virulence factors of group B Streptococcus isolated from invasive patients at Arabcare Hospital Laboratory, Palestine. Am. J. Infect. Control 2022, 50, 934–940. [Google Scholar] [CrossRef]
- Furfaro, L.L.; Chang, B.J.; Payne, M.S.; Chang, B.J.; Payne, M.S. Perinatal Streptococcus agalactiae Epidemiology and Surveillance Targets. Clin. Microbiol. Rev. 2018, 31, e00049-18. [Google Scholar] [CrossRef] [Green Version]
- Shabayek, S.; Spellerberg, B. Group B Streptococcal Colonization, Molecular Characteristics, and Epidemiology. Front. Microbiol. 2018, 9, 437. [Google Scholar] [CrossRef]
- Perme, T.; Golparian, D.; Bombek Ihan, M.; Rojnik, A.; Lucovnik, M.; Kornhauser Cerar, L.; Fister, P.; Lozar Krivec, J.; Grosek, S.; Ihan, A.; et al. Genomic and phenotypic characterisation of invasive neonatal and colonising group B Streptococcus isolates from Slovenia, 2001–2018. BMC Infect. Dis. 2020, 20, 958. [Google Scholar] [CrossRef]
- Munch-Petersen, E.; Christie, R.; Simmons, R.T.; Beddome, H.A. Further notes on a lytic phenomenon shown by group B streptococci. Aust. J. Exp. Biol. Med. Sci. 1945, 23, 193–195. [Google Scholar] [CrossRef] [PubMed]
- Jurgens, D.; Sterzik, B.; Fehrenbach, F.J. Unspecific binding of group B streptococcal cocytolysin (CAMP factor) to immunoglobulins and its possible role in pathogenicity. J. Exp. Med. 1987, 165, 720–732. [Google Scholar] [CrossRef] [PubMed]
- Ballard, M.B.; Mercado-Evans, V.; Marunde, M.G.; Nwanosike, H.; Zulk, J.; Patras, K.A. Group B Streptococcus CAMP Factor Does Not Contribute to Interactions with the Vaginal Epithelium and Is Dispensable for Vaginal Colonization in Mice. Microbiol. Spectr. 2021, 9, e0105821. [Google Scholar] [CrossRef] [PubMed]
- Hensler, M.E.; Quach, D.; Hsieh, C.J.; Doran, K.S.; Nizet, V. CAMP factor is not essential for systemic virulence of Group B Streptococcus. Microb. Pathog. 2008, 44, 84–88. [Google Scholar] [CrossRef] [PubMed]
- Carey, A.J.; Tan, C.K.; Mirza, S.; Irving-Rodgers, H.; Webb, R.I.; Lam, A.; Ulett, G.C. Infection and cellular defense dynamics in a novel 17beta-estradiol murine model of chronic human group B streptococcus genital tract colonization reveal a role for hemolysin in persistence and neutrophil accumulation. J. Immunol. 2014, 192, 1718–1731. [Google Scholar] [CrossRef] [Green Version]
- Liu, G.Y.; Doran, K.S.; Lawrence, T.; Turkson, N.; Puliti, M.; Tissi, L.; Nizet, V. Sword and shield: Linked group B streptococcal beta-hemolysin/cytolysin and carotenoid pigment function to subvert host phagocyte defense. Proc. Natl. Acad. Sci. USA 2004, 101, 14491–14496. [Google Scholar] [CrossRef] [Green Version]
- Tenenbaum, T.; Spellerberg, B.; Adam, R.; Vogel, M.; Kim, K.S.; Schroten, H. Streptococcus agalactiae invasion of human brain microvascular endothelial cells is promoted by the laminin-binding protein Lmb. Microbes Infect. 2007, 9, 714–720. [Google Scholar] [CrossRef]
- Pritchard, D.G.; Lin, B.; Willingham, T.R.; Baker, J.R. Characterization of the group B streptococcal hyaluronate lyase. Arch. Biochem. Biophys. 1994, 315, 431–437. [Google Scholar] [CrossRef]
- Davies, H.G.; Carreras-Abad, C.; Le Doare, K.; Heath, P.T. Group B Streptococcus: Trials and Tribulations. Pediatr. Infect. Dis. J. 2019, 38 (Suppl. S1), S72–S76. [Google Scholar] [CrossRef]
- Absalon, J.; Segall, N.; Block, S.L.; Center, K.J.; Scully, I.L.; Giardina, P.C.; Peterson, J.; Watson, W.J.; Gruber, W.C.; Jansen, K.U.; et al. Safety and immunogenicity of a novel hexavalent group B streptococcus conjugate vaccine in healthy, non-pregnant adults: A phase 1/2, randomised, placebo-controlled, observer-blinded, dose-escalation trial. Lancet Infect. Dis. 2021, 21, 263–274. [Google Scholar] [CrossRef]
- Duke, J.A.; Paschall, A.V.; Robinson, L.S.; Knoot, C.J.; Vinogradov, E.; Scott, N.E.; Feldman, M.F.; Avci, F.Y.; Harding, C.M. Development and Immunogenicity of a Prototype Multivalent Group B Streptococcus Bioconjugate Vaccine. ACS Infect. Dis. 2021, 7, 3111–3123. [Google Scholar] [CrossRef] [PubMed]
- Brokaw, A.; Nguyen, S.; Quach, P.; Orvis, A.; Furuta, A.; Johansson-Lindbom, B.; Fischer, P.B.; Rajagopal, L. A Recombinant Alpha-Like Protein Subunit Vaccine (GBS-NN) Provides Protection in Murine Models of Group B Streptococcus Infection. J. Infect. Dis. 2022, 226, 177–187. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.M.; Zhi, Y.; Ahn, K.B.; Lim, S.; Seo, H.S. Status of group B streptococcal vaccine development. Clin. Exp. Vaccine Res. 2018, 7, 76–81. [Google Scholar] [CrossRef] [PubMed]
- Chen, V.L.; Avci, F.Y.; Kasper, D.L. A maternal vaccine against group B Streptococcus: Past, present, and future. Vaccine 2013, 31 (Suppl. S4), D13–D19. [Google Scholar] [CrossRef]
- Baker, C.J.; Edwards, M.S. Group B streptococcal conjugate vaccines. Arch. Dis. Child. 2003, 88, 375–378. [Google Scholar] [CrossRef] [Green Version]
- Madhi, S.A.; Koen, A.; Cutland, C.L.; Jose, L.; Govender, N.; Wittke, F.; Olugbosi, M.; Sobanjo-Ter Meulen, A.; Baker, S.; Dull, P.M.; et al. Antibody Kinetics and Response to Routine Vaccinations in Infants Born to Women Who Received an Investigational Trivalent Group B Streptococcus Polysaccharide CRM197-Conjugate Vaccine during Pregnancy. Clin. Infect. Dis. 2017, 65, 1897–1904. [Google Scholar] [CrossRef]
- Song, J.Y.; Lim, J.H.; Lim, S.; Yong, Z.; Seo, H.S. Progress toward a group B streptococcal vaccine. Hum. Vaccin. Immunother. 2018, 14, 2669–2681. [Google Scholar] [CrossRef]
- Areschoug, T.; Stalhammar-Carlemalm, M.; Larsson, C.; Lindahl, G. Group B streptococcal surface proteins as targets for protective antibodies: Identification of two novel proteins in strains of serotype V. Infect. Immun. 1999, 67, 6350–6357. [Google Scholar] [CrossRef] [Green Version]
- Fischer, P.; Pawlowski, A.; Cao, D.; Bell, D.; Kitson, G.; Darsley, M.; Johansson-Lindbom, B. Safety and immunogenicity of a prototype recombinant alpha-like protein subunit vaccine (GBS-NN) against Group B Streptococcus in a randomised placebo-controlled double-blind phase 1 trial in healthy adult women. Vaccine 2021, 39, 4489–4499. [Google Scholar] [CrossRef]


{kind=link}
| Organization | Year | Guidelines | Important General Principles |
|---|---|---|---|
| CDC | 1996 | Prevention of perinatal group B streptococcal disease: a public health perspective. | Using one of two prevention strategies is recommended (details in [7]). |
| CDC, AAP, and ACOG | 1996 | Recommendations for intrapartum prophylaxis to prevent perinatal GBS disease (consensus guidelines) | To prevent early onset disease (EOD), intrapartum antibiotic prophylaxis (IAP) is recommended for pregnant women using antenatal cultures or a risk-factor-based approach. |
| CDC | 2002 | Prevention of perinatal group B streptococcal disease: revised guidelines from CDC. | Universal culture-based screening is recommended for all pregnant women at 35–37 weeks’ gestation to identify women who require intrapartum antibiotic prophylaxis (IAP), as well as a neonatal management algorithm for secondary prevention of GBS EOD |
| CDC | 2010 | Prevention of perinatal group B streptococcal disease: 2010 revision | Selection of specific antibiotics for the use of IAP in women in preterm labor and women with premature rupture of membranes. |
| AAP | 2019 | Prevention and management of perinatal GBS disease | Recommend that infants less than, greater than, or equal to 35 weeks’ gestation should be considered separately when performing risk assessment for GBS. |
| ACOG | 2020 | Prevention and management of perinatal GBS disease | Adjusting the start of antenatal antibiotic prophylaxis for pregnant women from 36 0/7 to 37 6/7 weeks of gestation. |
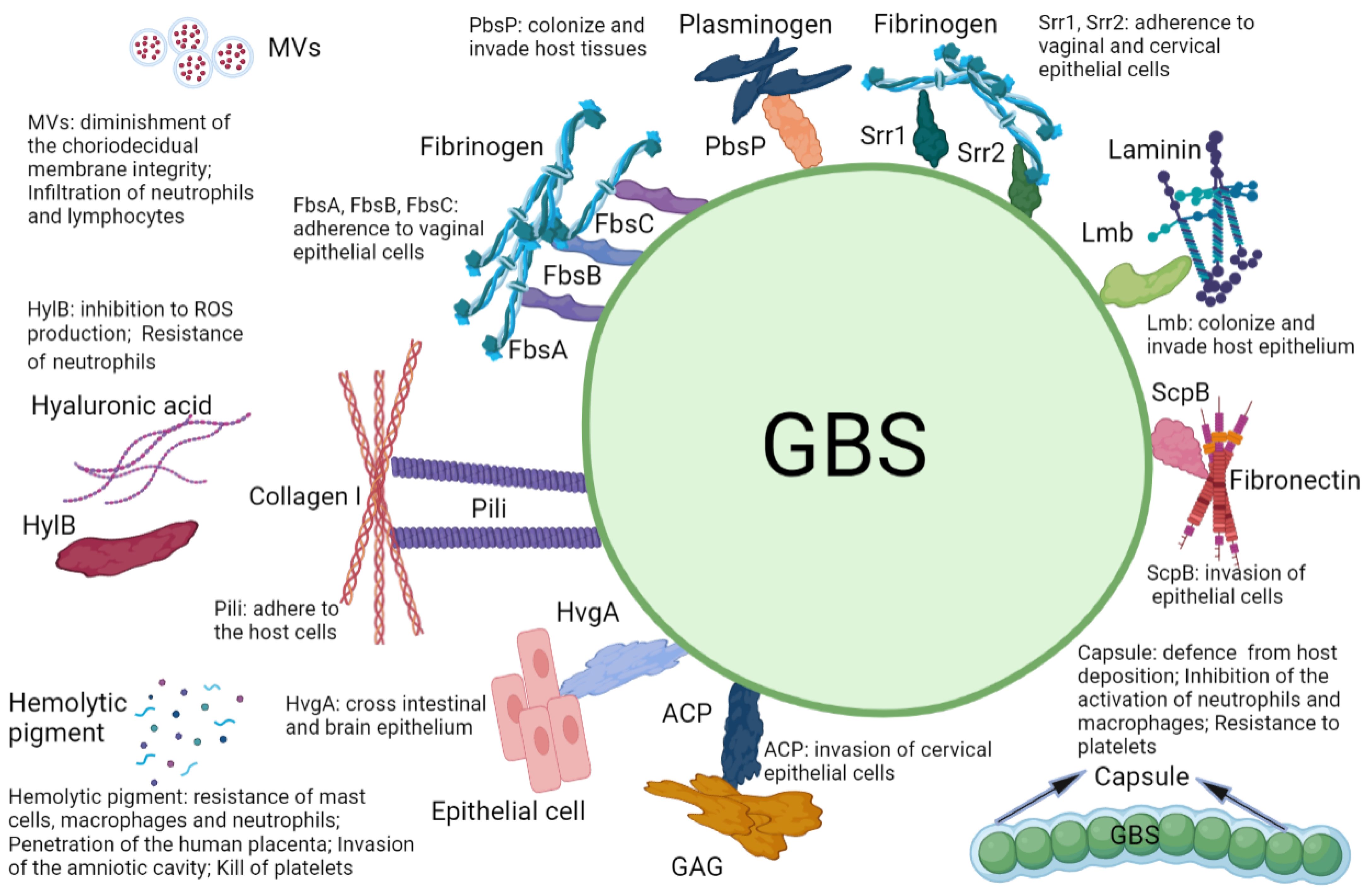
| Virulence Factor | Specific Target | Mechanism | Prevalence | Reference |
|---|---|---|---|---|
| Serine-rich repeats1 (Srr1) | Fibrinogen | Adherence to vaginal epithelial cells, cervical epithelial cells | Common | [15,25,89] |
| Serine-rich repeats2 (Srr2) | Fibrinogen, Plasmin, Plasminogen | Adherence to vaginal epithelial cells, cervical epithelial cells | Common | [15,25,89] |
| Fibrinogen-binding proteins (FbsA, FbsB, FbsC) | Fibrinogen | Adherence to vaginal epithelial cells | Common | [34,89] |
| Hypervirulent GBS adhesin (HvgA) | Unknown | Cross intestinal epithelium, brain epithelium | Common | [16,17,89] |
| Plasminogen binding surface protein (PbsP) | Plasminogen | Colonize and invade host tissues | Unknown | [37] |
| Pili | Collagen I | Adherence to host cells | Common | [28,43,89,95,96] |
| Laminin binding protein (Lmb) | Laminin | Colonize and invade host epithelium | Common | [47,89,97] |
| C5a peptidase (ScpB) | C5a, Fibronectin | Invasion of epithelial cells | Common | [50,51,89] |
| GBS hyaluronidase (HylB) | Hyaluronic acid | Inhibition to ROS production; resistance to neutrophils | Common | [56,58,98] |
| Hemolytic pigment | Neutrophils, Mast cells, Macrophages, Platelets | Resistance to mast cells, macrophages, and neutrophils; penetration of the human placenta; invasion of the amniotic cavity; kills platelets | Common | [18,19,63,65] |
| Alpha C protein (ACP) | Glycosaminog-lycans (GAG) | Invasion of cervical epithelial cells | Unknown | [66] |
| Capsule | Siglecs | Defense from host deposition; inhibition of the activation of neutrophils and macrophages; resistance to platelets | Common | [70,71,74,75,76] |
| Membrane vesicles (MVs) | Collagen | Diminishment of the choriodecidual membrane integrity; infiltration of neutrophils and lymphocytes | Common | [81,82] |
| Vaccine Candidate | Basic Components | Clinical Trial | Reference |
|---|---|---|---|
| Native polysaccharide vaccines | Capsule polysaccharide (CPS) | Ineffective | [103] |
| GBS glycoconjugate vaccine | Capsule polysaccharide (CPS) | Phase I | [104,105] |
| Trivalent (Ia, Ib, and III) CRM197 conjugate vaccine | Capsule polysaccharide (CPS) | Phase Ib/II | [106] |
| Pentavalent conjugate vaccine (Ia, Ib, II, III, and V) | Capsule polysaccharide (CPS) | Phase I/II | [107] |
| Hexavalent vaccines (Ia, Ib, II, III, IV, V) | Capsule polysaccharide (CPS) | Phase I/II | [99] |
| Prototype recombinant alpha-like protein subunit vaccine (GBS-NN) | Highly immunogenic N-terminal domains of Alpha C and Rib (GBS-NN) | Phase I | [99,108,109] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Y.; Liu, J. Group B Streptococcus: Virulence Factors and Pathogenic Mechanism. Microorganisms 2022, 10, 2483. https://doi.org/10.3390/microorganisms10122483
Liu Y, Liu J. Group B Streptococcus: Virulence Factors and Pathogenic Mechanism. Microorganisms. 2022; 10(12):2483. https://doi.org/10.3390/microorganisms10122483
Chicago/Turabian StyleLiu, Yuxin, and Jinhui Liu. 2022. "Group B Streptococcus: Virulence Factors and Pathogenic Mechanism" Microorganisms 10, no. 12: 2483. https://doi.org/10.3390/microorganisms10122483