

Microbiome Analysis of New, Insidious Cave Wall Alterations in the Apse of Lascaux Cave
 , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
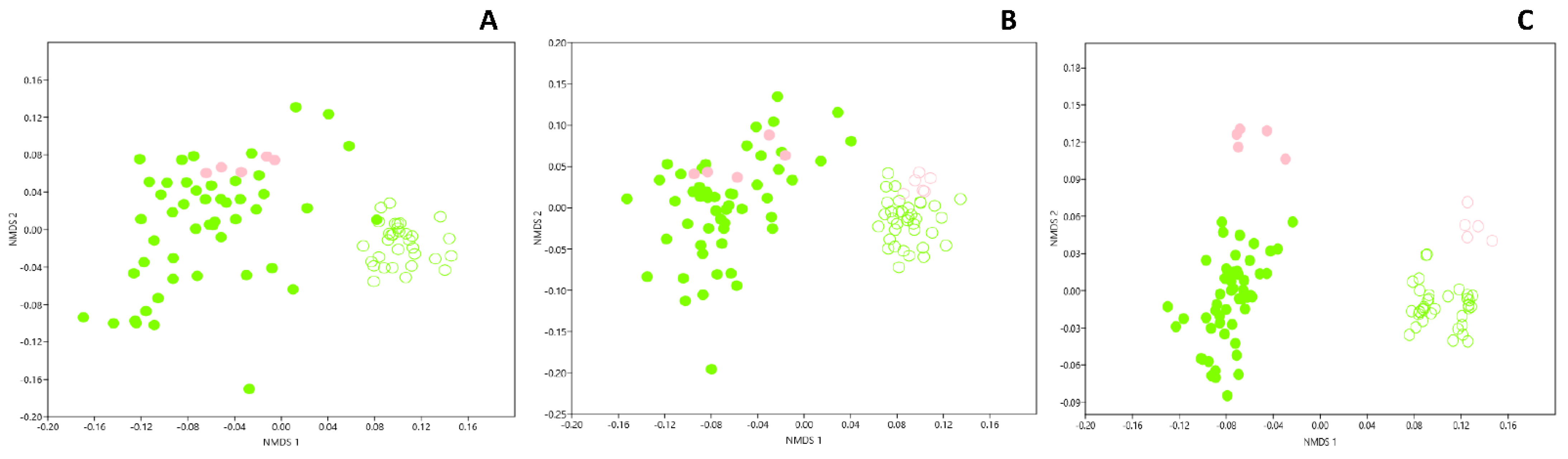{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sampling
2.2. DNA Extraction, Identification of Collembola and Illumina Sequencing
2.3. Bioinformatic Treatment of Illumina Sequence Data
2.4. Statistical Analyses
3. Results
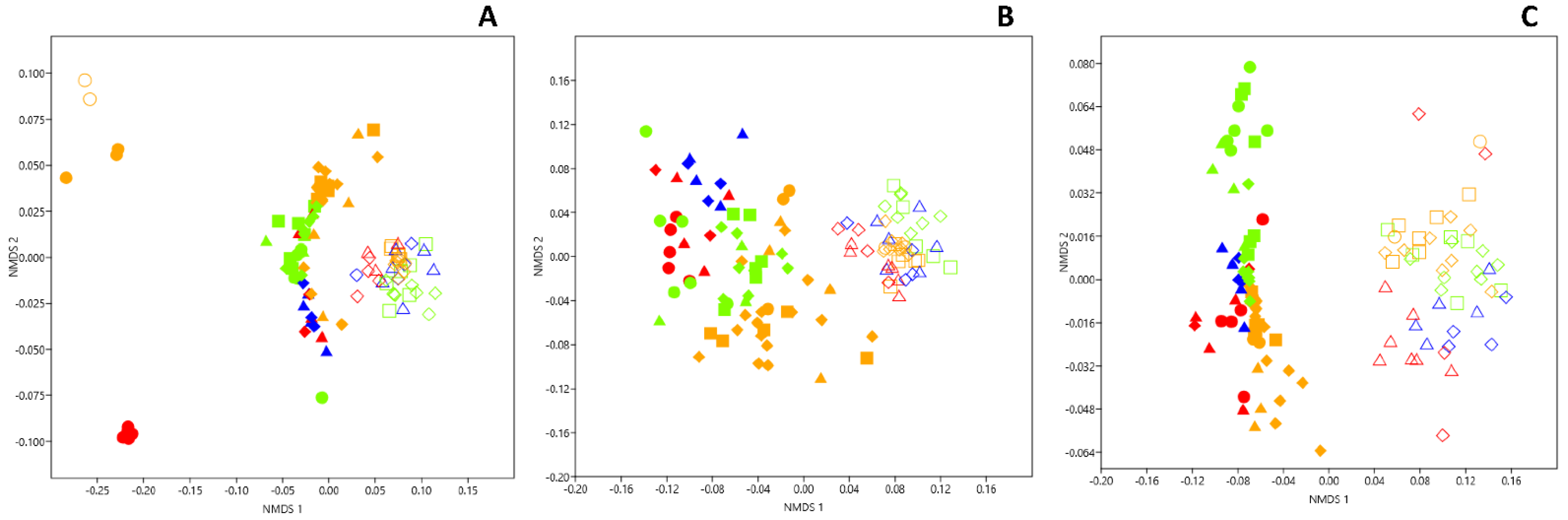
3.1. Genetic Structure of Fungal Community in Dark Zones
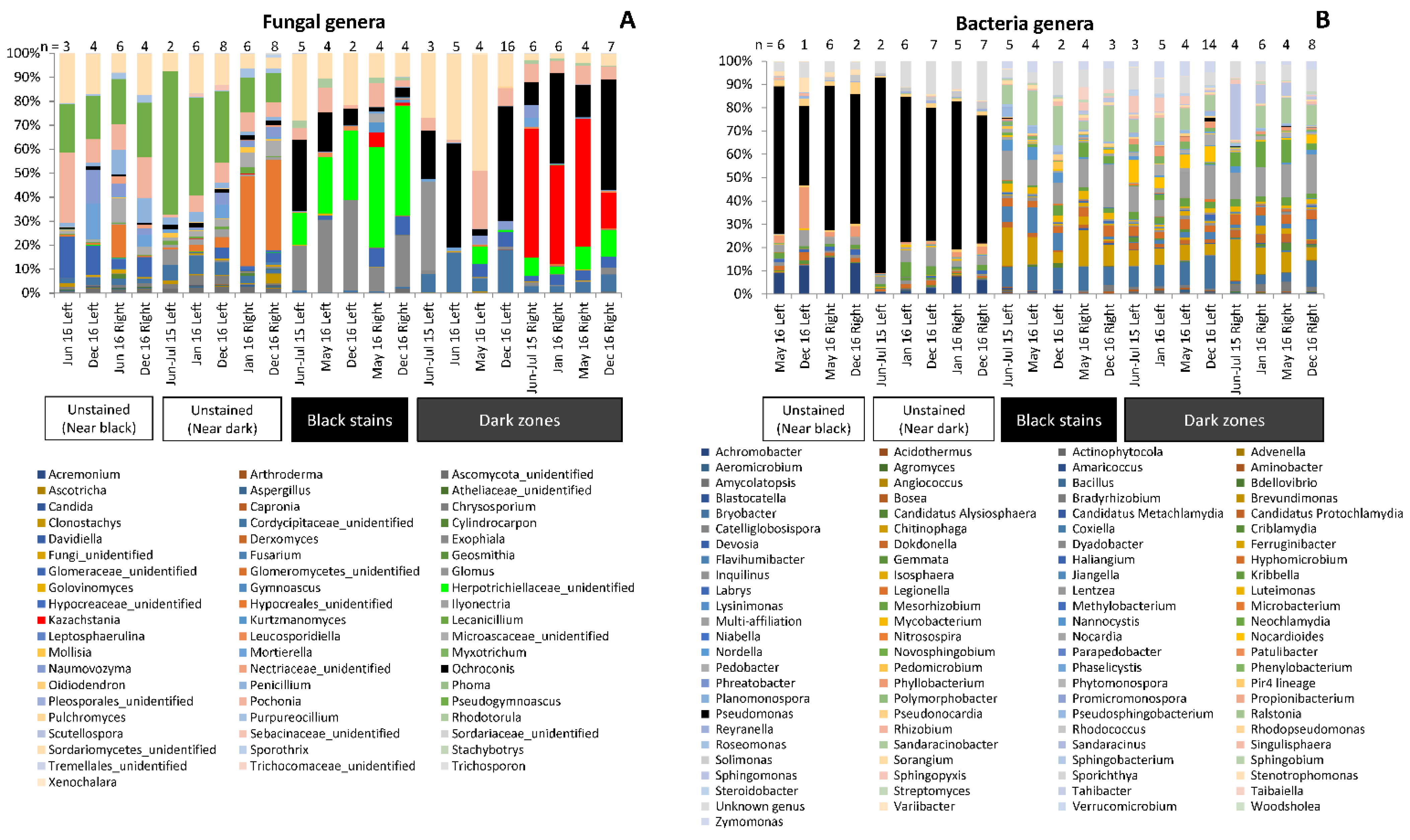
3.2. Fungal Community Composition in Dark Zones
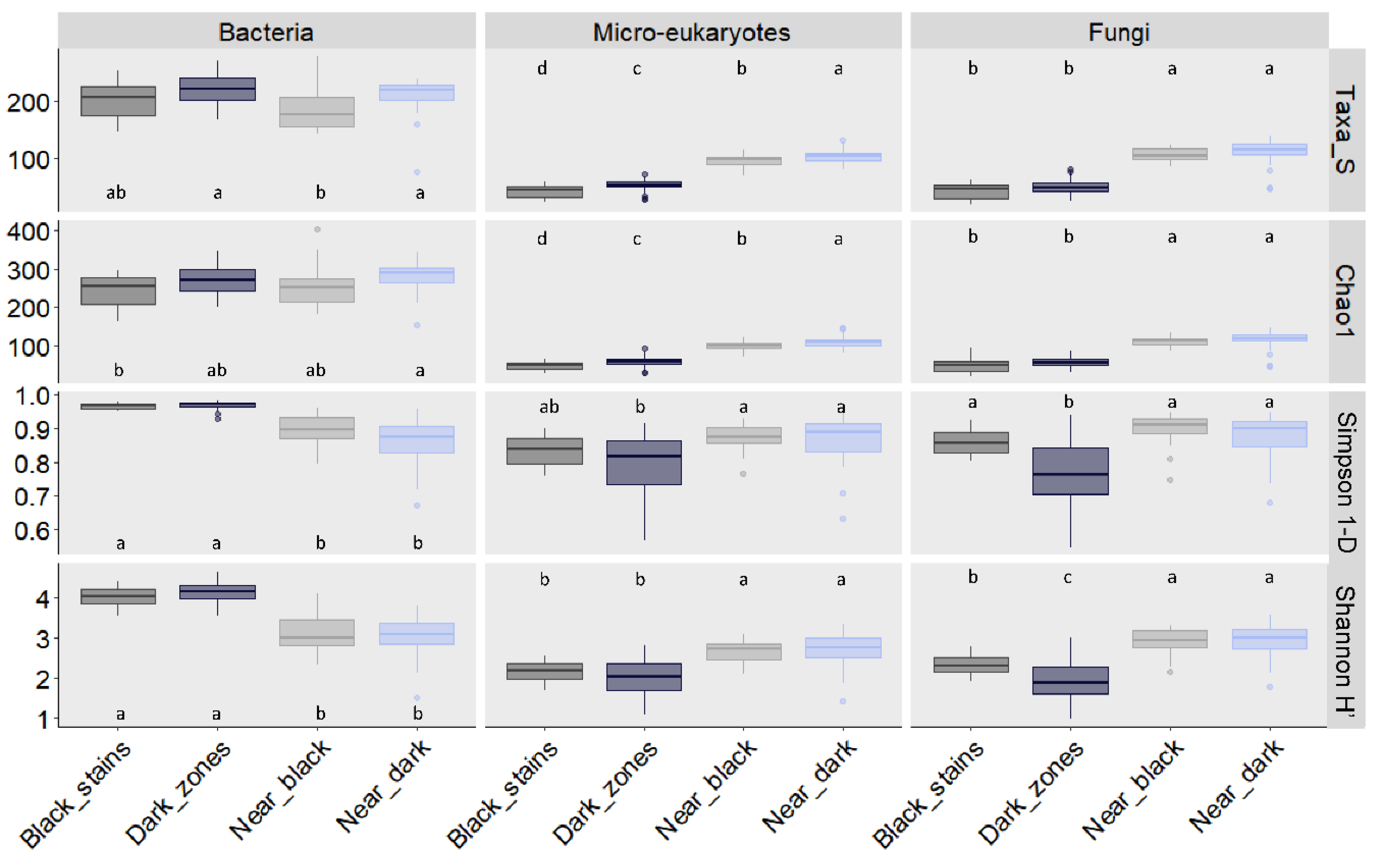
3.3. Fungal Community Size and Diversity in Dark Zones
3.4. Genetic Structure of Bacterial Community in Dark Zones
3.5. Bacterial Community Composition in Dark Zones
3.6. Bacterial Community Size and Diversity in Dark Zones
3.7. Microbial Comparison of Dark Zones in the Apse and the Nave
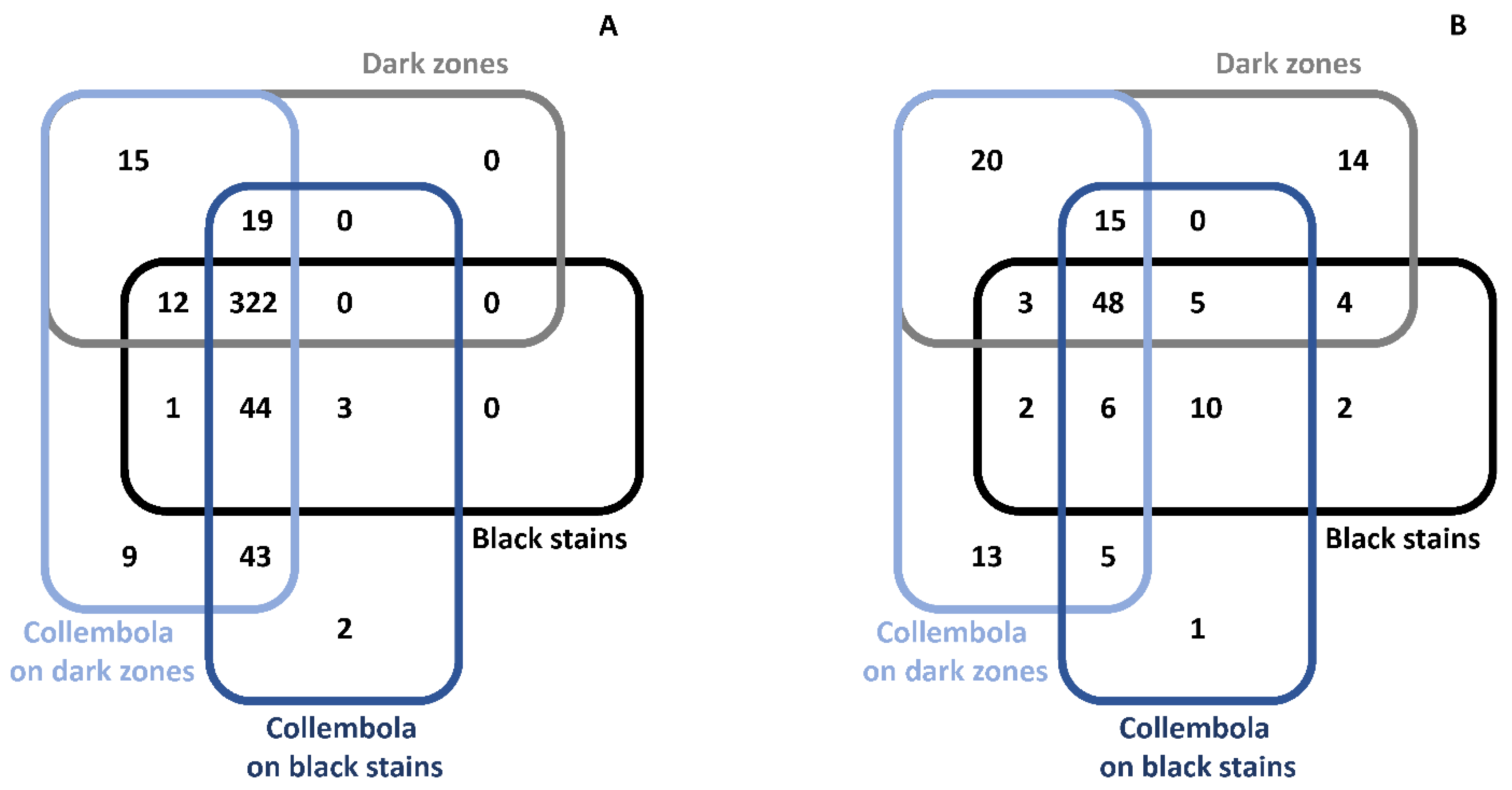
3.8. Microbiota Associated with Collembola Present on Dark Zones
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Schabereiter-Gurtner, C.; Saiz-Jimenez, C.; Piñar, G.; Lubitz, W.; Rölleke, S. Altamira cave Paleolithic paintings harbor partly unknown bacterial communities. FEMS Microbiol. Lett. 2002, 211, 7–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dupont, J.; Jacquet, C.; Dennetière, B.; Lacoste, S.; Bousta, F.; Orial, G.; Cruaud, C.; Couloux, A.; Roquebert, M.F. Invasion of the French Paleolithic painted cave of Lascaux by members of the Fusarium solani species complex. Mycologia 2007, 99, 526–533. [Google Scholar] [CrossRef] [PubMed]
- Alonso, L.; Pommier, T.; Kaufmann, B.; Dubost, A.; Chapulliot, D.; Doré, J.; Douady, C.J.; Moënne-Loccoz, Y. Anthropization level of Lascaux Cave microbiome shown by regional-scale comparisons of pristine and anthropized caves. Mol. Ecol. 2019, 28, 3383–3394. [Google Scholar] [CrossRef] [PubMed]
- Bastian, F.; Jurado, V.; Novakova, A.; Alabouvette, C.; Sáiz-Jiménez, C. The microbiology of Lascaux cave. Microbiology 2010, 156, 644–652. [Google Scholar] [CrossRef] [Green Version]
- Wei, T.P.; Zhang, H.; Zeng, X.Y.; Crous, P.W.; Jiang, Y.L. Re-evaluation of Sympoventuriaceae. Pers.-Mol. Phylogeny Evol. Fungi 2022, 48, 219–260. [Google Scholar] [CrossRef]
- Shen, M.; Zhang, J.Q.; Zhao, L.L.; Groenewald, J.Z.; Crous, P.W.; Zhang, Y. Venturiales. Stud. Mycol. 2020, 96, 185–308. [Google Scholar] [CrossRef]
- Martin-Sanchez, P.M.; Nováková, A.; Bastian, F.; Alabouvette, C.; Saiz-Jimenez, C. Two new species of the genus Ochroconis, O. lascauxensis and O. anomala isolated from black stains in Lascaux Cave, France. Fungal Biol. 2012, 116, 574–589. [Google Scholar] [CrossRef]
- Martin-Sanchez, P.M.; Sanchez-Cortes, S.; Lopez-Tobar, E.; Jurado, V.; Bastian, F.; Alabouvette, C.; Saiz-Jimenez, C. The nature of black stains in Lascaux Cave, France, as revealed by surface-enhanced Raman spectroscopy. J. Raman. Spectrosc. 2012, 43, 464–467. [Google Scholar] [CrossRef] [Green Version]
- De la Rosa, J.M.; Martin-Sanchez, P.M.; Sanchez-Cortes, S.; Hermosin, B.; Knicker, H.; Saiz-Jimenez, C. Structure of melanins from the fungi Ochroconis lascauxensis and Ochroconis anomala contaminating rock art in the Lascaux Cave. Sci. Rep. 2017, 7, 13441. [Google Scholar] [CrossRef] [Green Version]
- Alonso, L.; Pommier, T.; Simon, L.; Maucourt, F.; Doré, J.; Dubost, A.; Trân Van, V.; Minard, G.; Valiente Moro, C.; Douady, C.J.; et al. Microbiome analysis in Lascaux Cave in relation to black stain alterations of rock surfaces and collembola. Environ. Microbiol. Rep. 2022, in press. [Google Scholar] [CrossRef]
- Folmer, O.; Black, M.; Hoeh, W.; Lutz, R.; Vrijenhoek, R. DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. Mol. Mar. Biol. Biotechnol. 1994, 3, 294–299. [Google Scholar] [PubMed]
- Johnson, M.; Zaretskaya, I.; Raytselis, Y.; Merezhuk, Y.; McGinnis, S.; Madden, T.L. NCBI BLAST: A better web interface. Nucleic Acids Res. 2008, 36, W5–W9. [Google Scholar] [CrossRef] [PubMed]
- Klindworth, A.; Pruesse, E.; Schweer, T.; Peplies, J.; Quast, C.; Horn, M.; Glöckner, F.O. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 2013, 41, e1. [Google Scholar] [CrossRef] [PubMed]
- Toju, H.; Tanabe, A.S.; Yamamoto, S.; Sato, H. High-coverage ITS primers for the DNA-based identification of Ascomycetes and Basidiomycetes in environmental samples. PLoS ONE 2012, 7, e40863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dollive, S.; Peterfreund, G.L.; Sherrill-Mix, S.; Bittinger, K.; Sinha, R.; Hoffmann, C.; Nabel, C.S.; Hill, D.A.; Artis, D.; Bachman, M.A.; et al. A tool kit for quantifying eukaryotic rRNA gene sequences from human microbiome samples. Genome Biol. 2012, 13, R60. [Google Scholar] [CrossRef] [Green Version]
- Magoc, T.; Salzberg, S.L. FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef] [Green Version]
- Mahé, F.; Rognes, T.; Quince, C.; de Vargas, C.; Dunthorn, M. Swarm: Robust and fast clustering method for amplicon-based studies. PeerJ 2014, 2, e593. [Google Scholar] [CrossRef] [Green Version]
- Rognes, T.; Flouri, T.; Nichols, B.; Quince, C.; Mahé, F. VSEARCH: A versatile open source tool for metagenomics. PeerJ 2016, 4, e2584. [Google Scholar] [CrossRef] [Green Version]
- Bokulich, N.A.; Subramanian, S.; Faith, J.J.; Gevers, D.; Gordon, J.I.; Knight, R.; Mills, D.A.; Caporaso, J.G. Quality-filtering vastly improves diversity estimates from Illumina amplicon sequencing. Nat. Methods 2013, 10, 57–59. [Google Scholar] [CrossRef]
- Lan, Y.; Wang, Q.; Cole, J.R.; Rosen, G.L. Using the RDP classifier to predict taxonomic novelty and reduce the search space for finding novel organisms. PLoS ONE 2012, 7, e32491. [Google Scholar] [CrossRef]
- Pruesse, E.; Quast, C.; Knittel, K.; Fuchs, B.M.; Ludwig, W.; Peplies, J.; Glöckner, F.O. SILVA: A comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res. 2007, 35, 7188–7196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kõljalg, U.; Nilsson, R.H.; Abarenkov, K.; Tedersoo, L.; Taylor, A.F.S.; Bahram, M.; Bates, S.T.; Bruns, T.D.; Bengtsson-Palme, J.; Callaghan, T.M.; et al. Towards a unified paradigm for sequence-based identification of fungi. Mol. Ecol. 2013, 22, 5271–5277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Escudié, F.; Auer, L.; Bernard, M.; Mariadassou, M.; Cauquil, L.; Vidal, K.; Maman, S.; Hernandez-Raquet, G.; Combes, S.; Pascal, G. FROGS: Find, Rapidly, OTUs with Galaxy Solution. Bioinformatics 2017, 34, 1287–1294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chao, A. Estimating the population size for capture-recapture data with unequal catchability. Biometrics 1987, 43, 783–791. [Google Scholar] [CrossRef] [PubMed]
- Shannon, C.E. A mathematical theory of communication. Bell Syst. Tech. J. 1948, 27, 623–656. [Google Scholar] [CrossRef]
- Simpson, E.H. Measurement of diversity. Nature 1949, 163, 688. [Google Scholar] [CrossRef]
- Hammer, Ø.; Harper, D.A.; Ryan, P.D. PAST: Paleontological statistics software package for education and data analysis. Palaeontol. Electron. 2001, 4, 9. [Google Scholar]
- Bray, J.R.; Curtis, J.T. An ordination of the upland forest communities of southern Wisconsin. Ecol. Monogr. 1957, 27, 325–349. [Google Scholar] [CrossRef]
- Clarke, K.R. Non-parametric multivariate analyses of changes in community structure. Austral. Ecol. 1993, 18, 117–143. [Google Scholar] [CrossRef]
- Bastian, F.; Alabouvette, C.; Saiz-Jimenez, C. The impact of arthropods on fungal community structure in Lascaux Cave. J. Appl. Microbiol. 2009, 106, 1456–1462. [Google Scholar] [CrossRef]
- Alonso, L.; Creuzé-des-Châtelliers, C.; Trabac, T.; Dubost, A.; Moënne-Loccoz, Y.; Pommier, T. Rock substrate rather than black stain alterations drives microbial community structure in the passage of Lascaux Cave. Microbiome 2018, 6, 216. [Google Scholar] [CrossRef] [PubMed]
- Hiscox, J.; Savoury, M.; Müller, C.T.; Lindahl, B.D.; Rogers, H.J.; Boddy, L. Priority effects during fungal community establishment in beech wood. ISME J. 2015, 9, 2246–2260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukami, T.; Dickie, I.A.; Wilkie, J.P.; Paulus, B.C.; Park, D.; Roberts, A.; Buchanan, P.K.; Allen, R.B. Assembly history dictates ecosystem functioning: Evidence from wood decomposer communities. Ecol. Lett. 2010, 13, 675–684. [Google Scholar] [CrossRef] [PubMed]
- Thimm, T.; Hoffmann, A.; Borkott, H.; Munch, J.C.; Tebbe, C.C. The gut of the soil microarthropod Folsomia candida (Collembola) is a frequently changeable but selective habitat and a vector for microorganisms. Appl. Environ. Microbiol. 1998, 64, 2660–2669. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alonso, L.; Pommier, T.; Abrouk, D.; Hugoni, M.; Tran Van, V.; Minard, G.; Valiente Moro, C.; Moënne-Loccoz, Y. Microbiome Analysis of New, Insidious Cave Wall Alterations in the Apse of Lascaux Cave. Microorganisms 2022, 10, 2449. https://doi.org/10.3390/microorganisms10122449
Alonso L, Pommier T, Abrouk D, Hugoni M, Tran Van V, Minard G, Valiente Moro C, Moënne-Loccoz Y. Microbiome Analysis of New, Insidious Cave Wall Alterations in the Apse of Lascaux Cave. Microorganisms. 2022; 10(12):2449. https://doi.org/10.3390/microorganisms10122449
Chicago/Turabian StyleAlonso, Lise, Thomas Pommier, Danis Abrouk, Mylène Hugoni, Van Tran Van, Guillaume Minard, Claire Valiente Moro, and Yvan Moënne-Loccoz. 2022. "Microbiome Analysis of New, Insidious Cave Wall Alterations in the Apse of Lascaux Cave" Microorganisms 10, no. 12: 2449. https://doi.org/10.3390/microorganisms10122449