
The Spatial Organization of Bacterial Transcriptional Regulatory Networks

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chromatin Interaction Data
2.2. Definition of Bins and Localization of Genes
2.3. Constructing 3D Models of Bacterial Chromosomes
2.4. Construction of Transcriptional Regulatory Networks
2.5. Determination of Network Hierarchy
2.6. Discovery of Network Motifs
3. Results
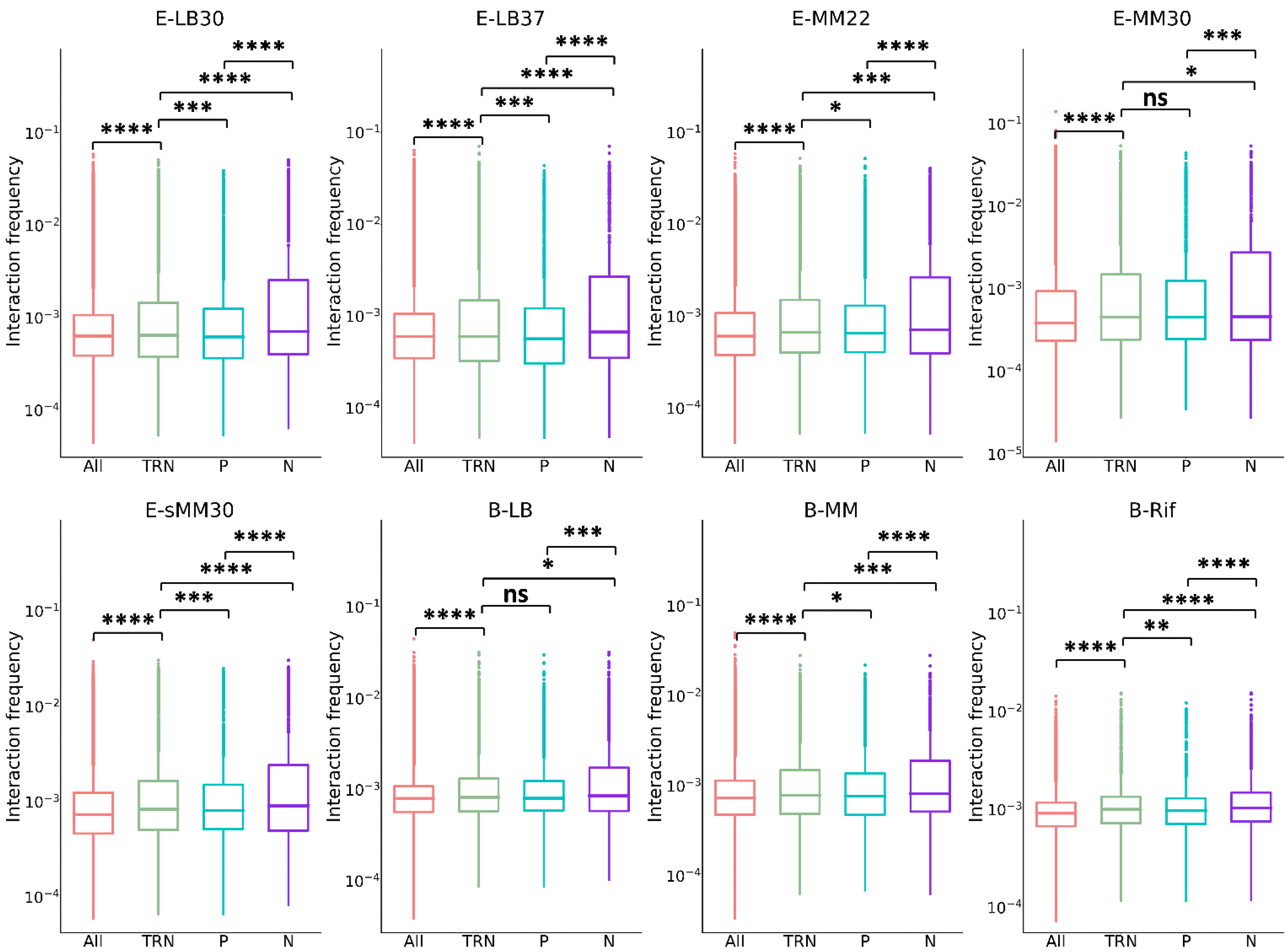
3.1. Chromatin Interaction within TRN
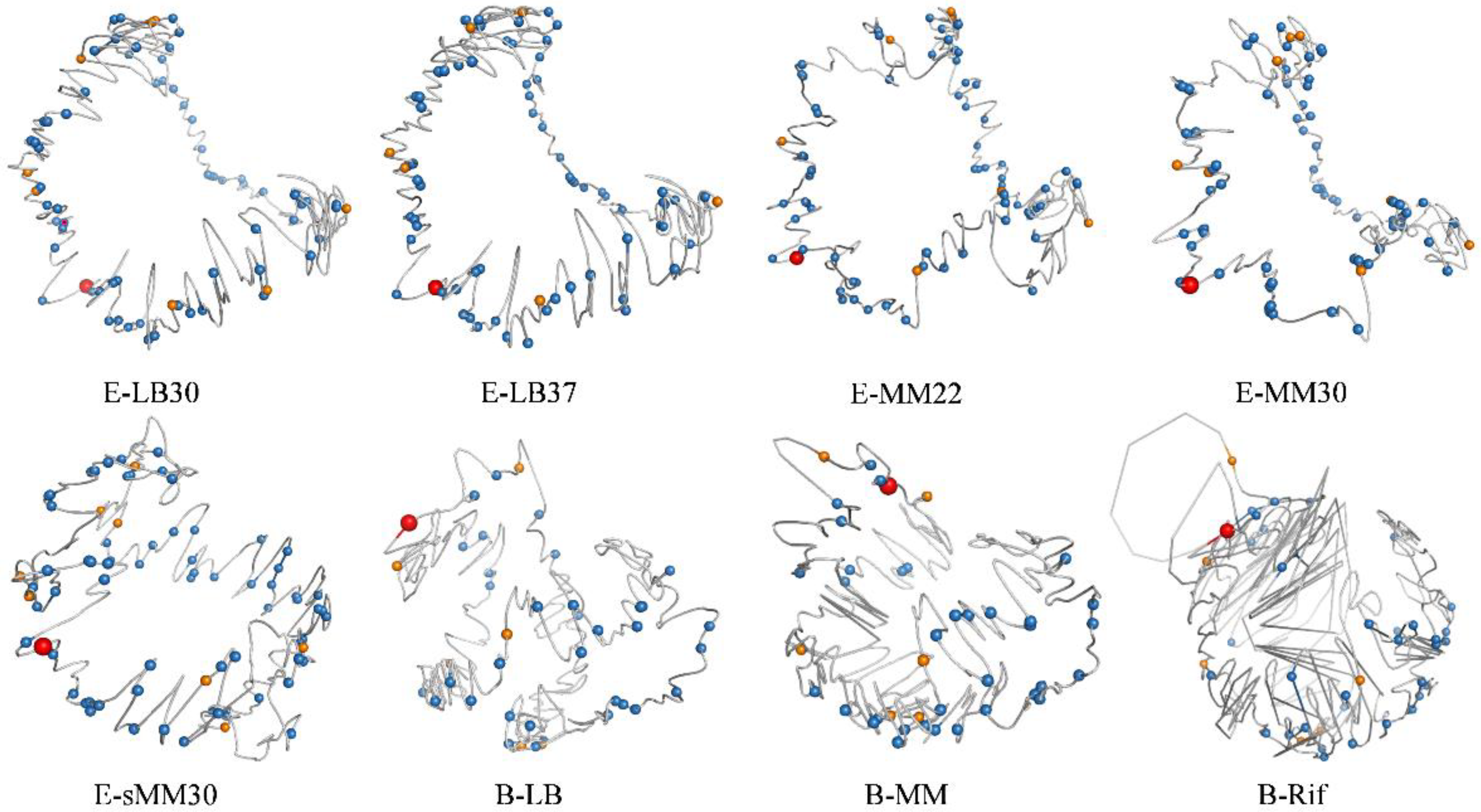
3.2. Chromatin Interaction of Central Nodes in TRN
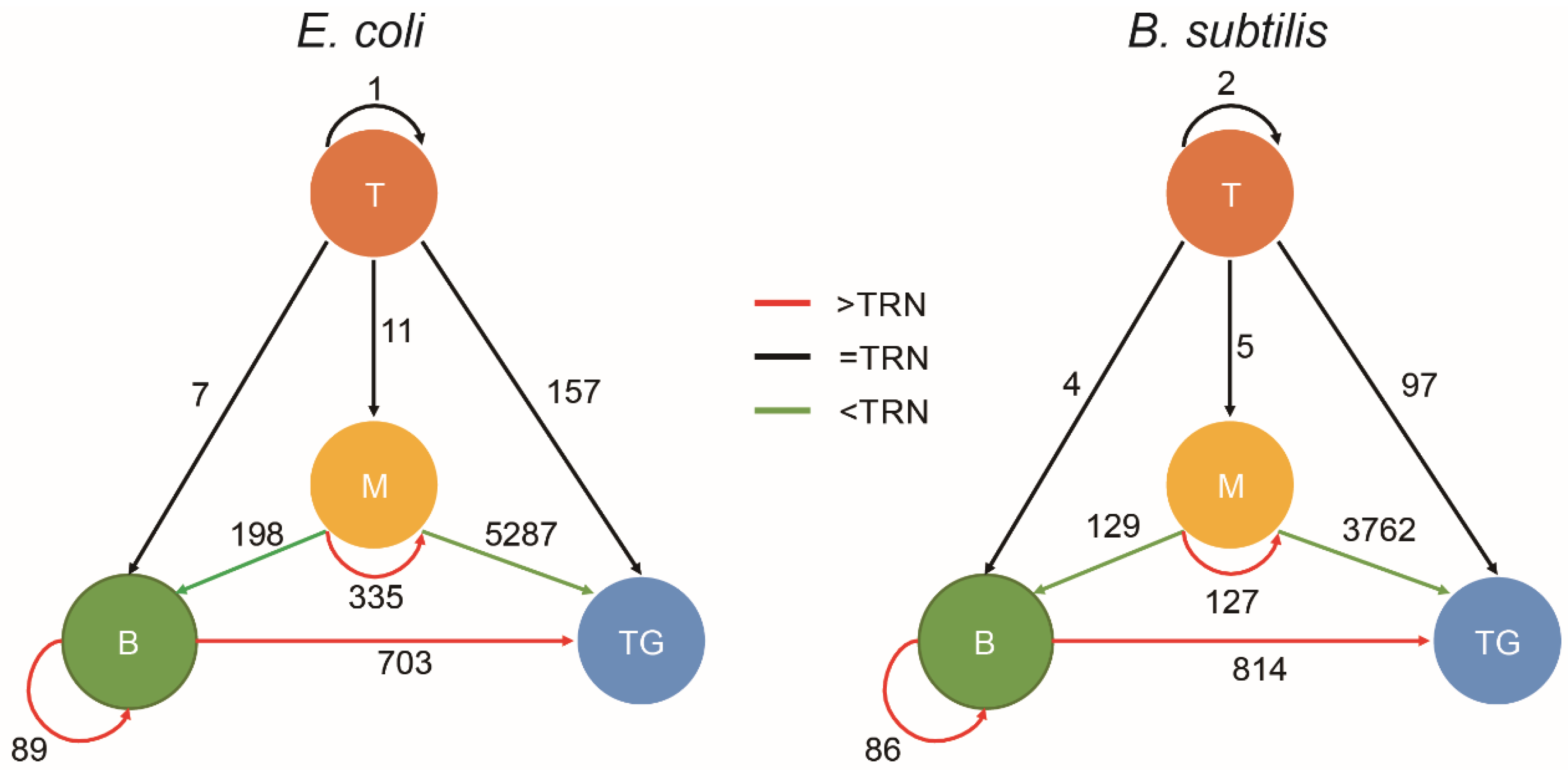
3.3. Chromatin Interaction in the Hierarchy of TRN
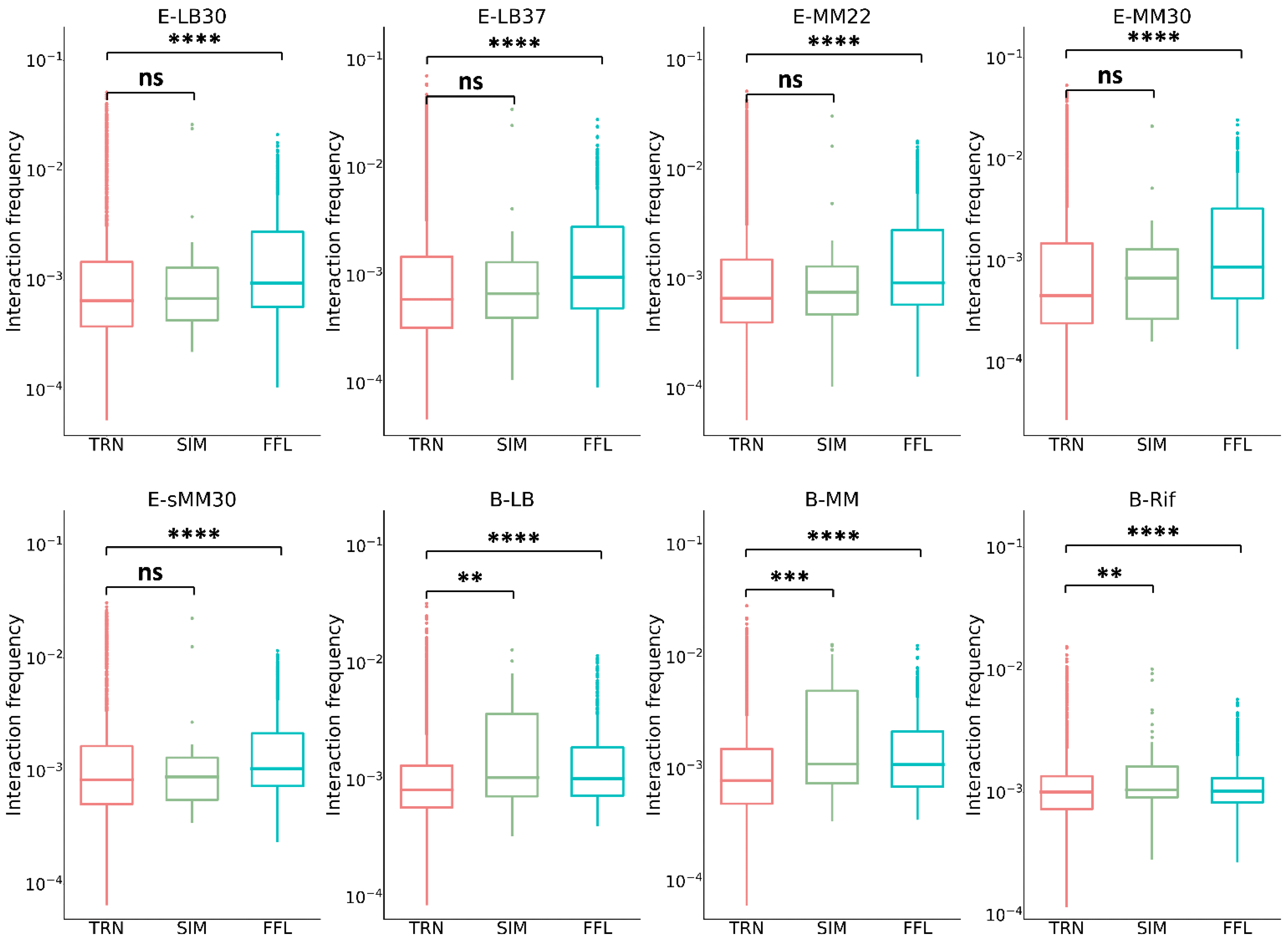
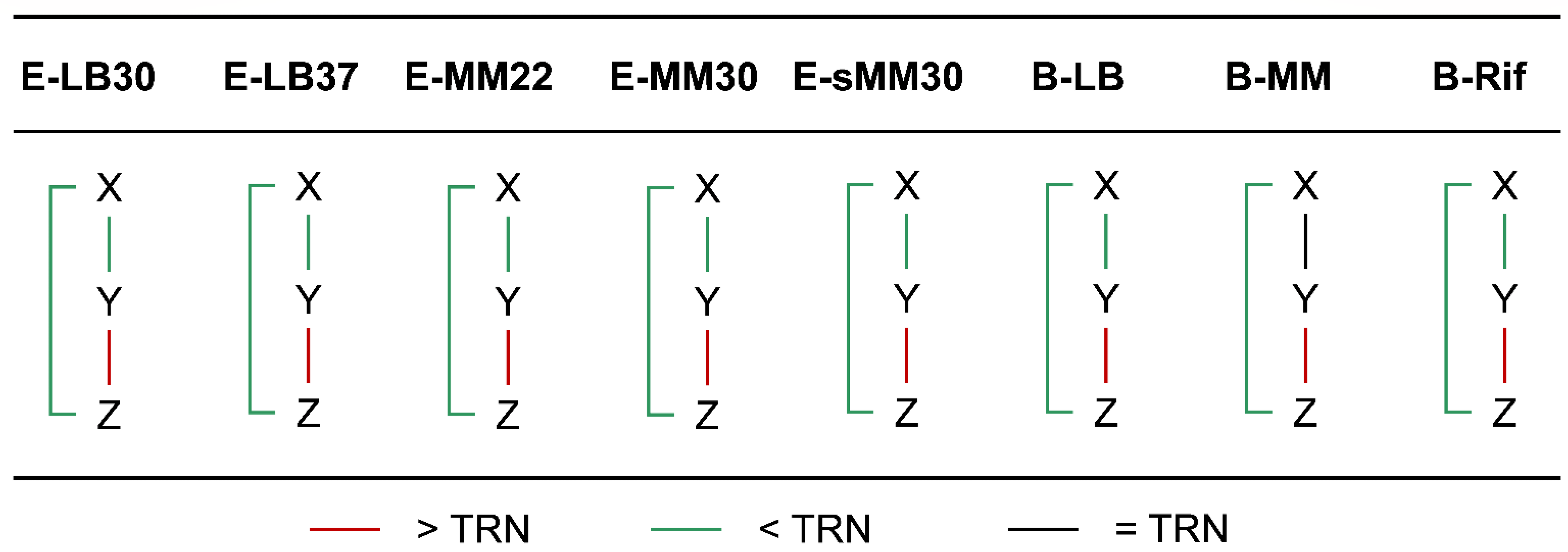
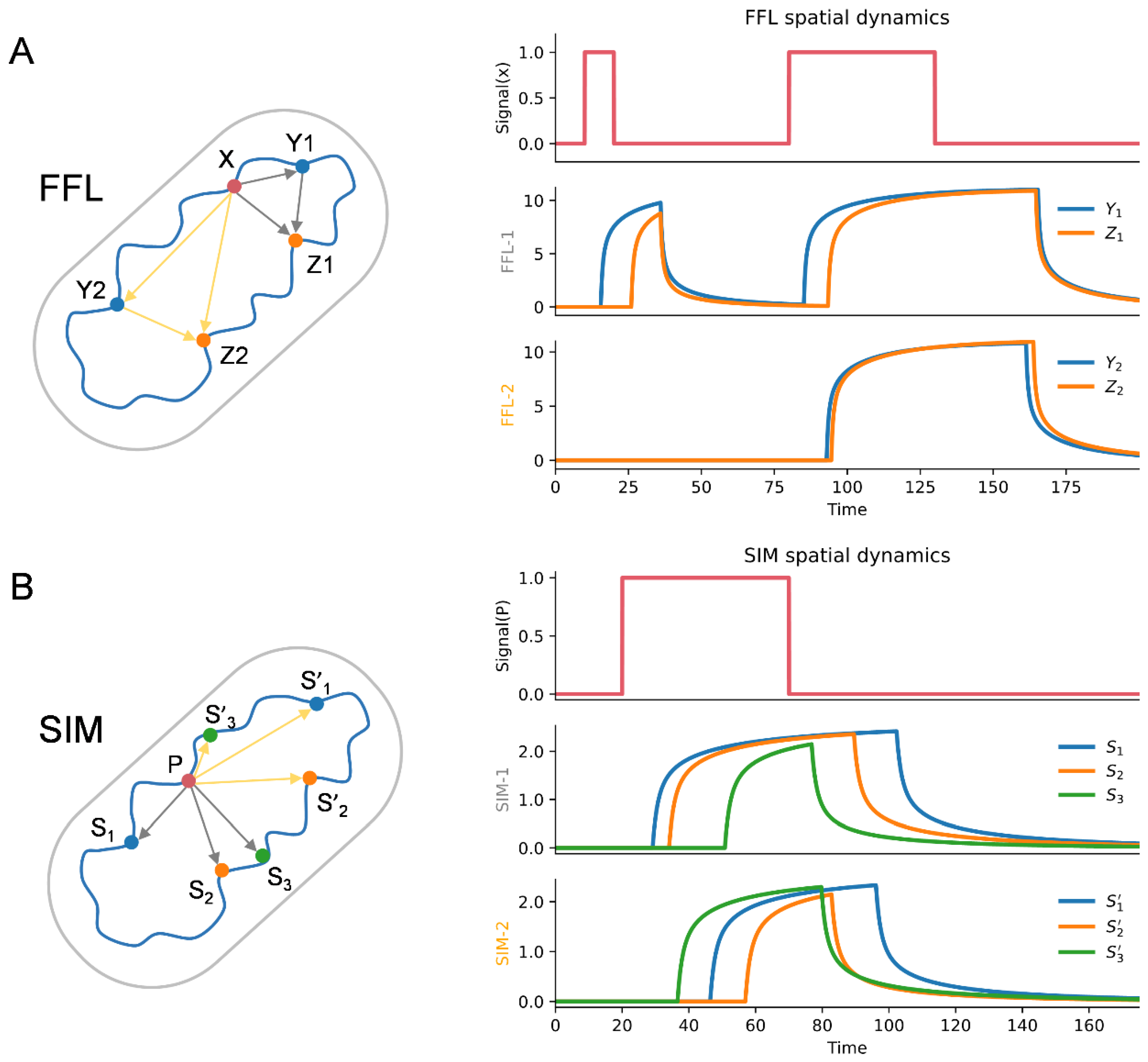
3.4. Chromatin Interaction in the Network Motifs of TRN
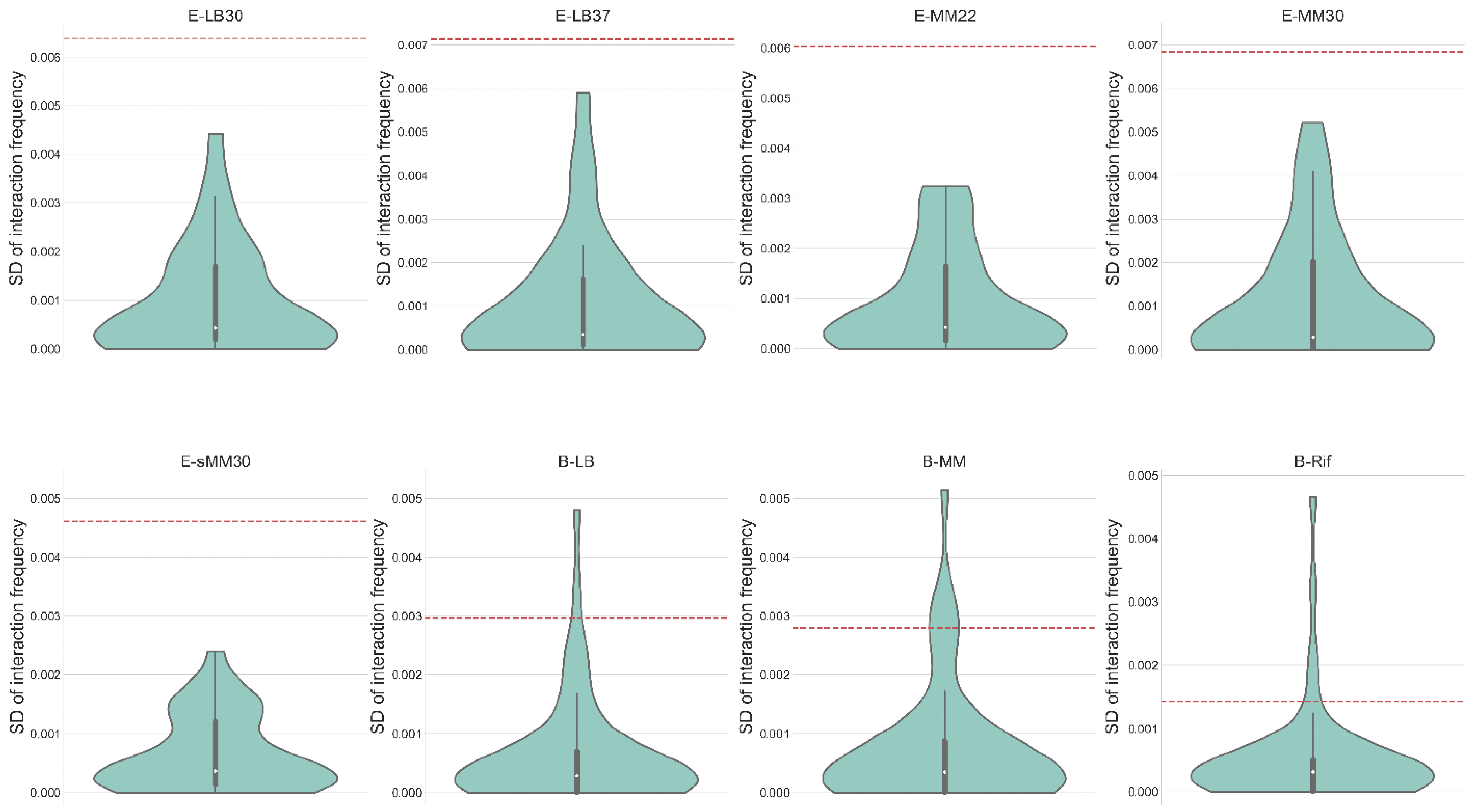
3.5. 3D Modeling of the Bacterial Chromosome Structures
3.6. Spatial Effect in the Dynamic Function of Network Motifs
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Balleza, E.; Lopez-Bojorquez, L.N.; Martinez-Antonio, A.; Resendis-Antonio, O.; Lozada-Chavez, I.; Balderas-Martinez, Y.I.; Encarnacion, S.; Collado-Vides, J. Regulation by transcription factors in bacteria: Beyond description. FEMS Microbiol. Rev. 2009, 33, 133–151. [Google Scholar] [CrossRef] [Green Version]
- Seshasayee, A.S.; Bertone, P.; Fraser, G.M.; Luscombe, N.M. Transcriptional regulatory networks in bacteria: From input signals to output responses. Curr. Opin. Microbiol. 2006, 9, 511–519. [Google Scholar] [CrossRef]
- Mejia-Almonte, C.; Busby, S.J.W.; Wade, J.T.; van Helden, J.; Arkin, A.P.; Stormo, G.D.; Eilbeck, K.; Palsson, B.O.; Galagan, J.E.; Collado-Vides, J. Redefining fundamental concepts of transcription initiation in bacteria. Nat. Rev. Genet. 2020, 21, 699–714. [Google Scholar] [CrossRef]
- Madan Babu, M.; Teichmann, S.A.; Aravind, L. Evolutionary dynamics of prokaryotic transcriptional regulatory networks. J. Mol. Biol. 2006, 358, 614–633. [Google Scholar] [CrossRef]
- Bhardwaj, N.; Kim, P.M.; Gerstein, M.B. Rewiring of transcriptional regulatory networks: Hierarchy, rather than connectivity, better reflects the importance of regulators. Sci. Signal. 2010, 3, ra79. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.; Gerstein, M. Genomic analysis of the hierarchical structure of regulatory networks. Proc. Natl. Acad. Sci. USA 2006, 103, 14724–14731. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Antonio, A.; Janga, S.C.; Thieffry, D. Functional organisation of Escherichia coli transcriptional regulatory network. J. Mol. Biol. 2008, 381, 238–247. [Google Scholar] [CrossRef] [Green Version]
- Resendis-Antonio, O.; Freyre-Gonzalez, J.A.; Menchaca-Mendez, R.; Gutierrez-Rios, R.M.; Martinez-Antonio, A.; Avila-Sanchez, C.; Collado-Vides, J. Modular analysis of the transcriptional regulatory network of E. coli. Trends Genet. TIG 2005, 21, 16–20. [Google Scholar] [CrossRef]
- Ma, H.W.; Buer, J.; Zeng, A.P. Hierarchical structure and modules in the Escherichia coli transcriptional regulatory network revealed by a new top-down approach. BMC Bioinform. 2004, 5, 199. [Google Scholar] [CrossRef] [Green Version]
- Alon, U. Network motifs: Theory and experimental approaches. Nat. Rev. Genet. 2007, 8, 450–461. [Google Scholar] [CrossRef]
- Andrews, S.S.; Arkin, A.P. Simulating cell biology. Curr. Biol. 2006, 16, R523–R527. [Google Scholar] [CrossRef] [Green Version]
- Hammar, P.; Leroy, P.; Mahmutovic, A.; Marklund, E.G.; Berg, O.G.; Elf, J. The lac repressor displays facilitated diffusion in living cells. Science 2012, 336, 1595–1598. [Google Scholar] [CrossRef]
- Jones, D.L.; Leroy, P.; Unoson, C.; Fange, D.; Curic, V.; Lawson, M.J.; Elf, J. Kinetics of dCas9 target search in Escherichia coli. Science 2017, 357, 1420–1423. [Google Scholar] [CrossRef]
- Pulkkinen, O.; Metzler, R. Distance matters: The impact of gene proximity in bacterial gene regulation. Phys. Rev. Lett. 2013, 110, 198101. [Google Scholar] [CrossRef] [Green Version]
- Mahmutovic, A.; Berg, O.G.; Elf, J. What matters for lac repressor search in vivo--sliding, hopping, intersegment transfer, crowding on DNA or recognition? Nucleic Acids Res. 2015, 43, 3454–3464. [Google Scholar] [CrossRef] [Green Version]
- Thevenin, A.; Ein-Dor, L.; Ozery-Flato, M.; Shamir, R. Functional gene groups are concentrated within chromosomes, among chromosomes and in the nuclear space of the human genome. Nucleic Acids Res. 2014, 42, 9854–9861. [Google Scholar] [CrossRef] [Green Version]
- Xie, T.; Fu, L.Y.; Yang, Q.Y.; Xiong, H.; Xu, H.; Ma, B.G.; Zhang, H.Y. Spatial features for Escherichia coli genome organization. BMC Genom. 2015, 16, 37. [Google Scholar] [CrossRef] [Green Version]
- Van, A.L.; Soula, H.A.; Berry, H. Space-induced bifurcation in repression-based transcriptional circuits. BMC Syst. Biol. 2014, 8, 125. [Google Scholar] [CrossRef] [Green Version]
- Weng, X.; Xiao, J. Spatial organization of transcription in bacterial cells. Trends Genet. TIG 2014, 30, 287–297. [Google Scholar] [CrossRef]
- Dame, R.T.; Rashid, F.M.; Grainger, D.C. Chromosome organization in bacteria: Mechanistic insights into genome structure and function. Nat. Rev. Genet. 2020, 21, 227–242. [Google Scholar] [CrossRef]
- Krogh, T.J.; Moller-Jensen, J.; Kaleta, C. Impact of Chromosomal Architecture on the Function and Evolution of Bacterial Genomes. Front. Microbiol. 2018, 9, 2019. [Google Scholar] [CrossRef]
- Hershberg, R.; Yeger-Lotem, E.; Margalit, H. Chromosomal organization is shaped by the transcription regulatory network. Trends Genet. TIG 2005, 21, 138–142. [Google Scholar] [CrossRef]
- Dorman, C.J. Genome architecture and global gene regulation in bacteria: Making progress towards a unified model? Nat. Rev. Microbiol. 2013, 11, 349–355. [Google Scholar] [CrossRef] [Green Version]
- Cagliero, C.; Grand, R.S.; Jones, M.B.; Jin, D.J.; O’Sullivan, J.M. Genome conformation capture reveals that the Escherichia coli chromosome is organized by replication and transcription. Nucleic Acids Res. 2013, 41, 6058–6071. [Google Scholar] [CrossRef] [Green Version]
- Lioy, V.S.; Cournac, A.; Marbouty, M.; Duigou, S.; Mozziconacci, J.; Espeli, O.; Boccard, F.; Koszul, R. Multiscale Structuring of the E. coli Chromosome by Nucleoid-Associated and Condensin Proteins. Cell 2018, 172, 771–783.e18. [Google Scholar] [CrossRef]
- Marbouty, M.; Le Gall, A.; Cattoni, D.I.; Cournac, A.; Koh, A.; Fiche, J.B.; Mozziconacci, J.; Murray, H.; Koszul, R.; Nollmann, M. Condensin- and Replication-Mediated Bacterial Chromosome Folding and Origin Condensation Revealed by Hi-C and Super-resolution Imaging. Mol. Cell 2015, 59, 588–602. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.D.; Le, T.B.K.; Lajoie, B.R.; Dekker, J.; Laub, M.T.; Rudner, D.Z. Condensin promotes the juxtaposition of DNA flanking its loading site in Bacillus subtilis. Gene Dev. 2015, 29, 1661–1675. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.D.; Brandao, H.B.; Le, T.B.K.; Laub, M.T.; Rudner, D.Z. Bacillus subtilis SMC complexes juxtapose chromosome arms as they travel from origin to terminus. Science 2017, 355, 524–527. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Montero Llopis, P.; Rudner, D.Z. Organization and segregation of bacterial chromosomes. Nat. Rev. Genet. 2013, 14, 191–203. [Google Scholar] [CrossRef] [Green Version]
- Cournac, A.; Marie-Nelly, H.; Marbouty, M.; Koszul, R.; Mozziconacci, J. Normalization of a chromosomal contact map. BMC Genom. 2012, 13, 436. [Google Scholar] [CrossRef]
- Hua, K.J.; Ma, B.G. EVR: Reconstruction of bacterial chromosome 3D structure models using error-vector resultant algorithm. BMC Genom. 2019, 20, 738. [Google Scholar] [CrossRef] [Green Version]
- Gama-Castro, S.; Salgado, H.; Santos-Zavaleta, A.; Ledezma-Tejeida, D.; Muniz-Rascado, L.; Garcia-Sotelo, J.S.; Alquicira-Hernandez, K.; Martinez-Flores, I.; Pannier, L.; Castro-Mondragon, J.A.; et al. RegulonDB version 9.0: High-level integration of gene regulation, coexpression, motif clustering and beyond. Nucleic Acids Res. 2016, 44, D133–D143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michna, R.H.; Zhu, B.Y.; Mader, U.; Stulke, J. SubtiWiki 2.0-an integrated database for the model organism Bacillus subtilis. Nucleic Acids Res. 2016, 44, D654–D662. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Vendruscolo, M.; Singh, A.; Kumar, D.; Samal, A. Analysis of the hierarchical structure of the B. subtilis transcriptional regulatory network. Mol. Biosyst. 2015, 11, 930–941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kashtan, N.; Itzkovitz, S.; Milo, R.; Alon, U. Efficient sampling algorithm for estimating subgraph concentrations and detecting network motifs. Bioinformatics 2004, 20, 1746–1758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konagurthu, A.S.; Lesk, A.M. Single and multiple input modules in regulatory networks. Proteins 2008, 73, 320–324. [Google Scholar] [CrossRef]
- Paget, M.S. Bacterial Sigma Factors and Anti-Sigma Factors: Structure, Function and Distribution. Biomolecules 2015, 5, 1245–1265. [Google Scholar] [CrossRef]
- Cook, P.R. A model for all genomes: The role of transcription factories. J. Mol. Biol. 2010, 395, 1–10. [Google Scholar] [CrossRef]
- Bonev, B.; Cavalli, G. Organization and function of the 3D genome. Nat. Rev. Genet. 2016, 17, 661–678. [Google Scholar] [CrossRef]
- Le Berre, D.; Reverchon, S.; Muskhelishvili, G.; Nasser, W. Relationship between the Chromosome Structural Dynamics and Gene Expression-A Chicken and Egg Dilemma? Microorganisms 2022, 10, 846. [Google Scholar] [CrossRef]
- Le, D.H.; Kwon, Y.K. A coherent feedforward loop design principle to sustain robustness of biological networks. Bioinformatics 2013, 29, 630–637. [Google Scholar] [CrossRef] [Green Version]
- Cakir, E.; Lesne, A.; Hutt, M.T. The economy of chromosomal distances in bacterial gene regulation. NPJ Syst. Biol. Appl. 2021, 7, 49. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Yin, Y.; Olman, V.; Xu, Y. Genomic arrangement of regulons in bacterial genomes. PLoS ONE 2012, 7, e29496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warren, P.B.; ten Wolde, P.R. Statistical analysis of the spatial distribution of operons in the transcriptional regulation network of Escherichia coli. J. Mol. Biol. 2004, 342, 1379–1390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, D.Q.; Tian, L.; Ma, B.G. Spatial organization of the transcriptional regulatory network of Saccharomyces cerevisiae. FEBS Lett. 2019, 593, 876–884. [Google Scholar] [CrossRef]
- Lagomarsino, M.C.; Espeli, O.; Junier, I. From structure to function of bacterial chromosomes: Evolutionary perspectives and ideas for new experiments. FEBS Lett. 2015, 589 Pt A, 2996–3004. [Google Scholar] [CrossRef] [Green Version]
- Deng, W.; Lee, J.; Wang, H.; Miller, J.; Reik, A.; Gregory, P.D.; Dean, A.; Blobel, G.A. Controlling long-range genomic interactions at a native locus by targeted tethering of a looping factor. Cell 2012, 149, 1233–1244. [Google Scholar] [CrossRef] [Green Version]
- Keung, A.J.; Joung, J.K.; Khalil, A.S.; Collins, J.J. Chromatin regulation at the frontier of synthetic biology. Nat. Rev. Genet. 2015, 16, 159–171. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tian, L.; Liu, T.; Hua, K.-J.; Hu, X.-P.; Ma, B.-G. The Spatial Organization of Bacterial Transcriptional Regulatory Networks. Microorganisms 2022, 10, 2366. https://doi.org/10.3390/microorganisms10122366
Tian L, Liu T, Hua K-J, Hu X-P, Ma B-G. The Spatial Organization of Bacterial Transcriptional Regulatory Networks. Microorganisms. 2022; 10(12):2366. https://doi.org/10.3390/microorganisms10122366
Chicago/Turabian StyleTian, Liu, Tong Liu, Kang-Jian Hua, Xiao-Pan Hu, and Bin-Guang Ma. 2022. "The Spatial Organization of Bacterial Transcriptional Regulatory Networks" Microorganisms 10, no. 12: 2366. https://doi.org/10.3390/microorganisms10122366