
Exploring Structural Diversity among Adhesion Devices Encoded by Lactococcal P335 Phages with AlphaFold2

Abstract
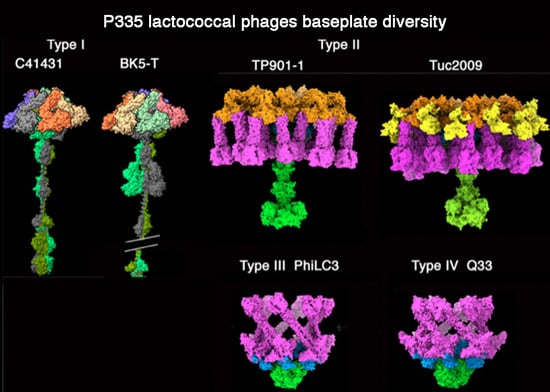
:
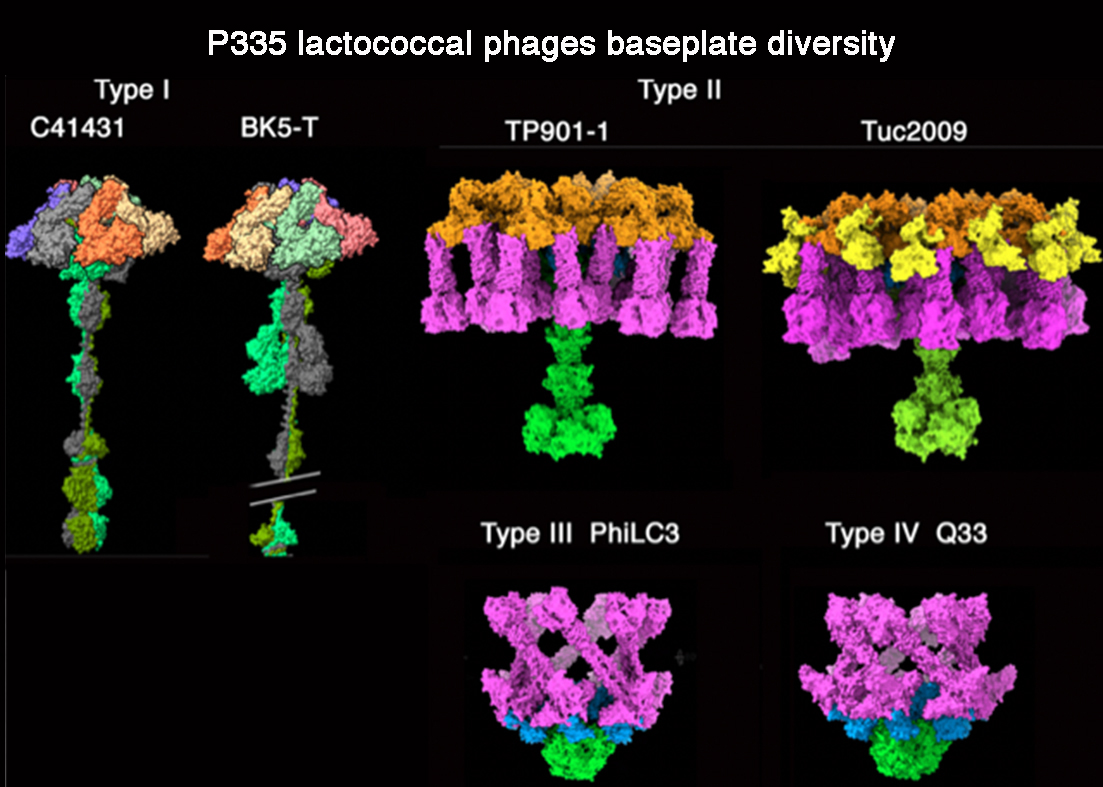{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
3. Results
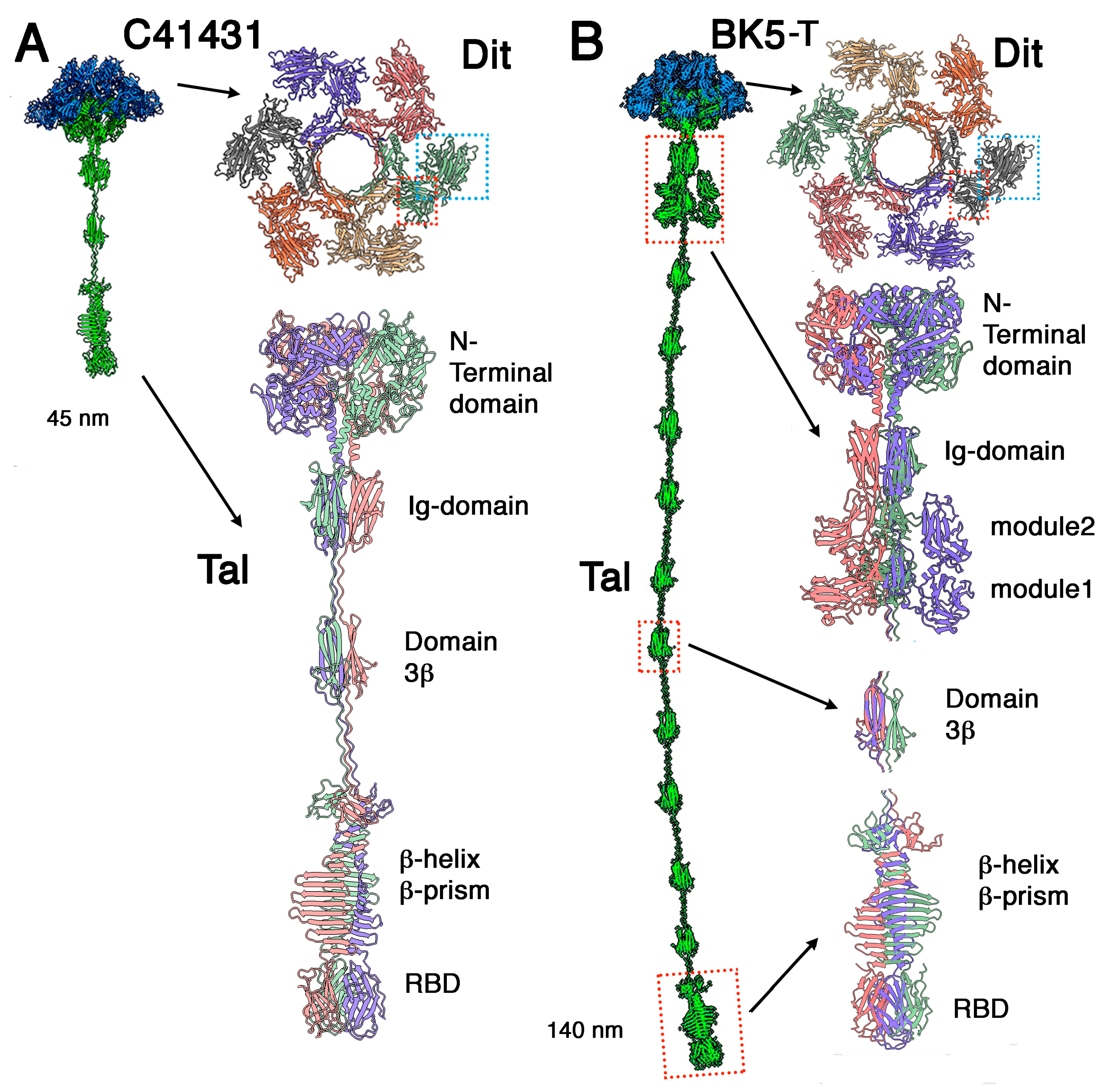
3.1. Phages Belonging to P335 Type I: C41431 and BK5-T
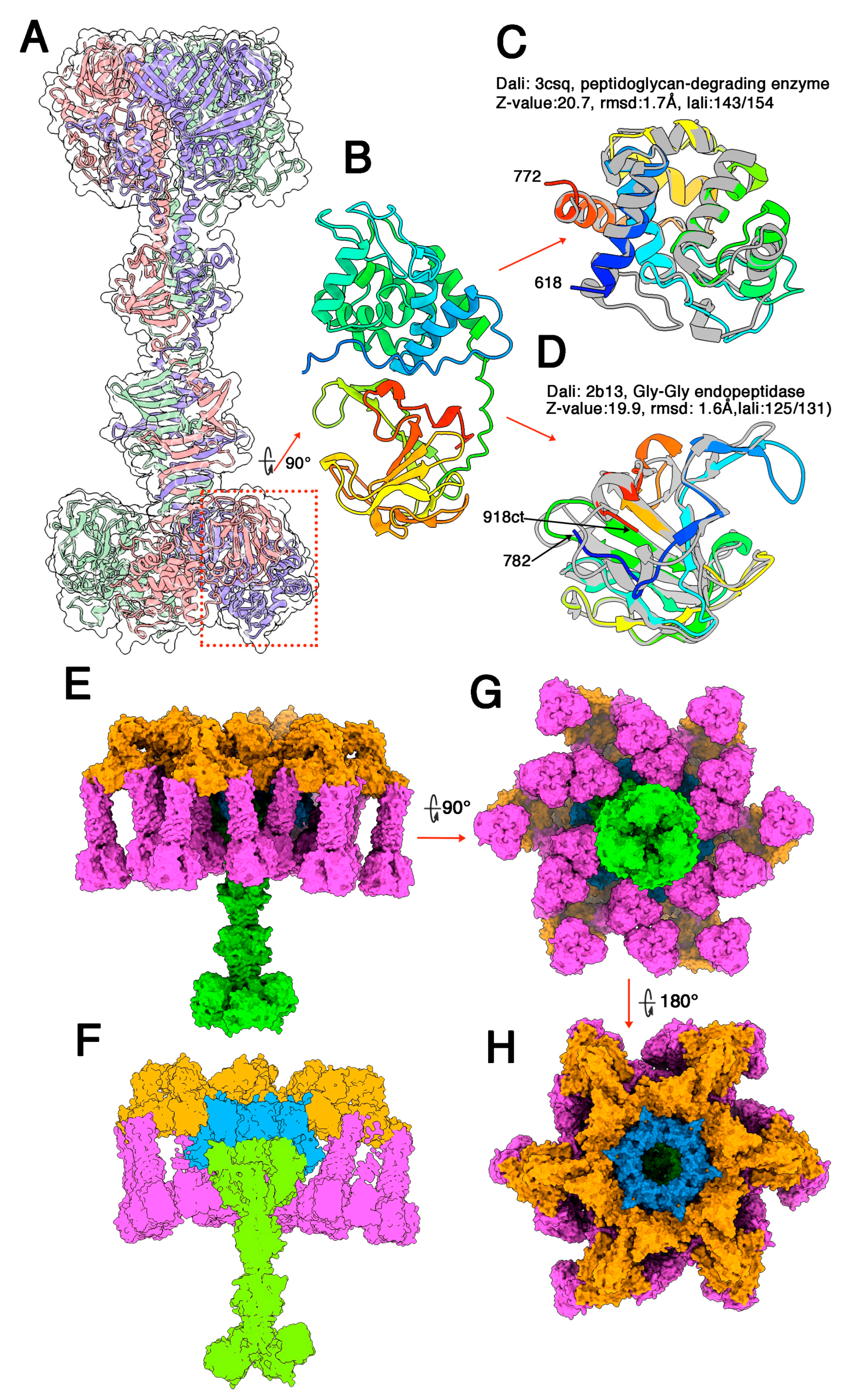
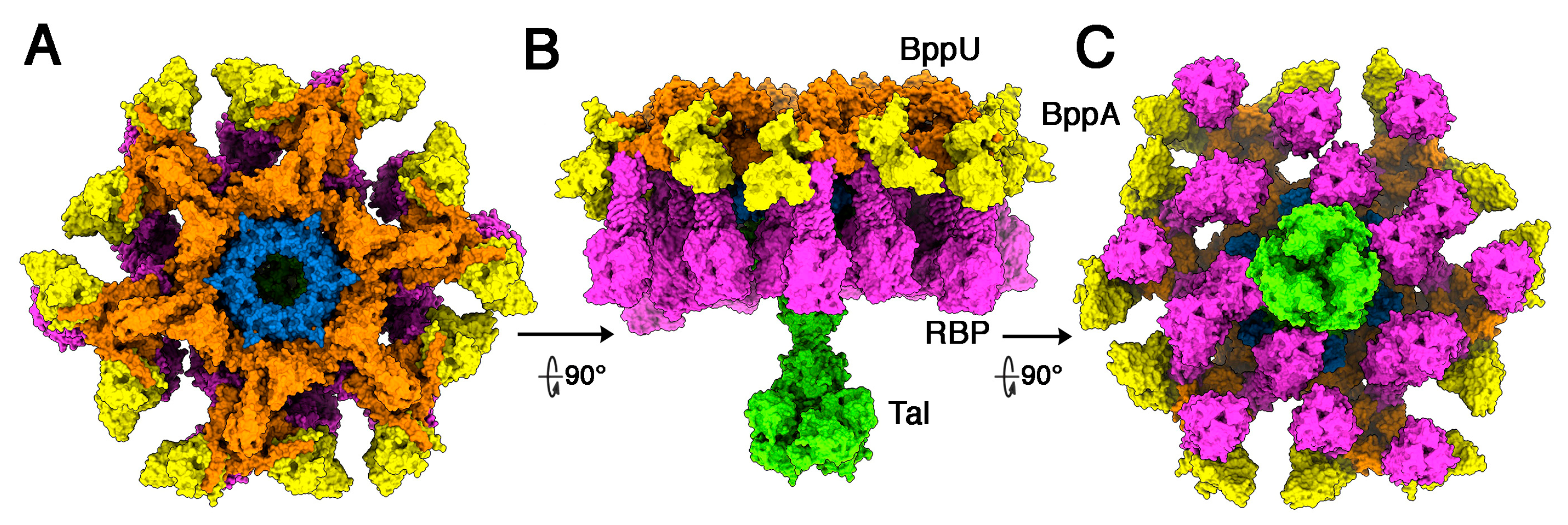
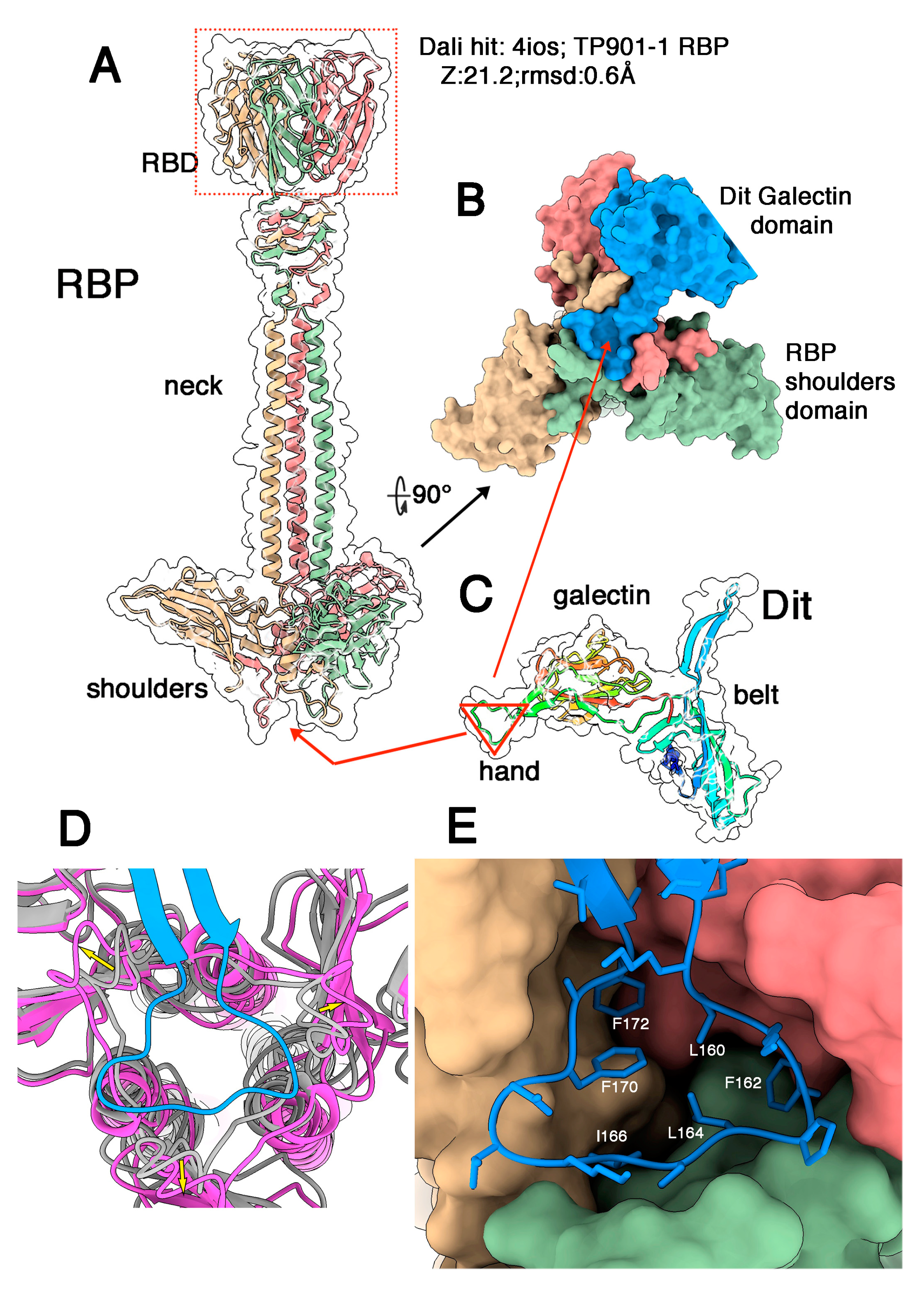
3.2. Phages Belonging to P335 Type II: TP901-1 and Tuc2009
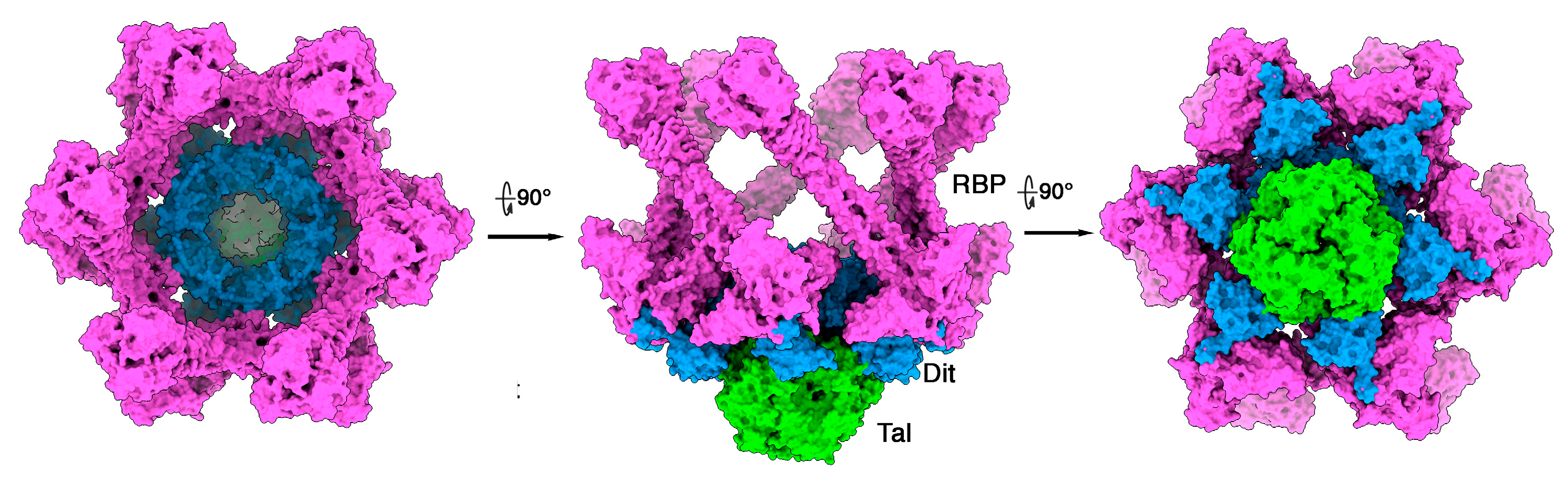
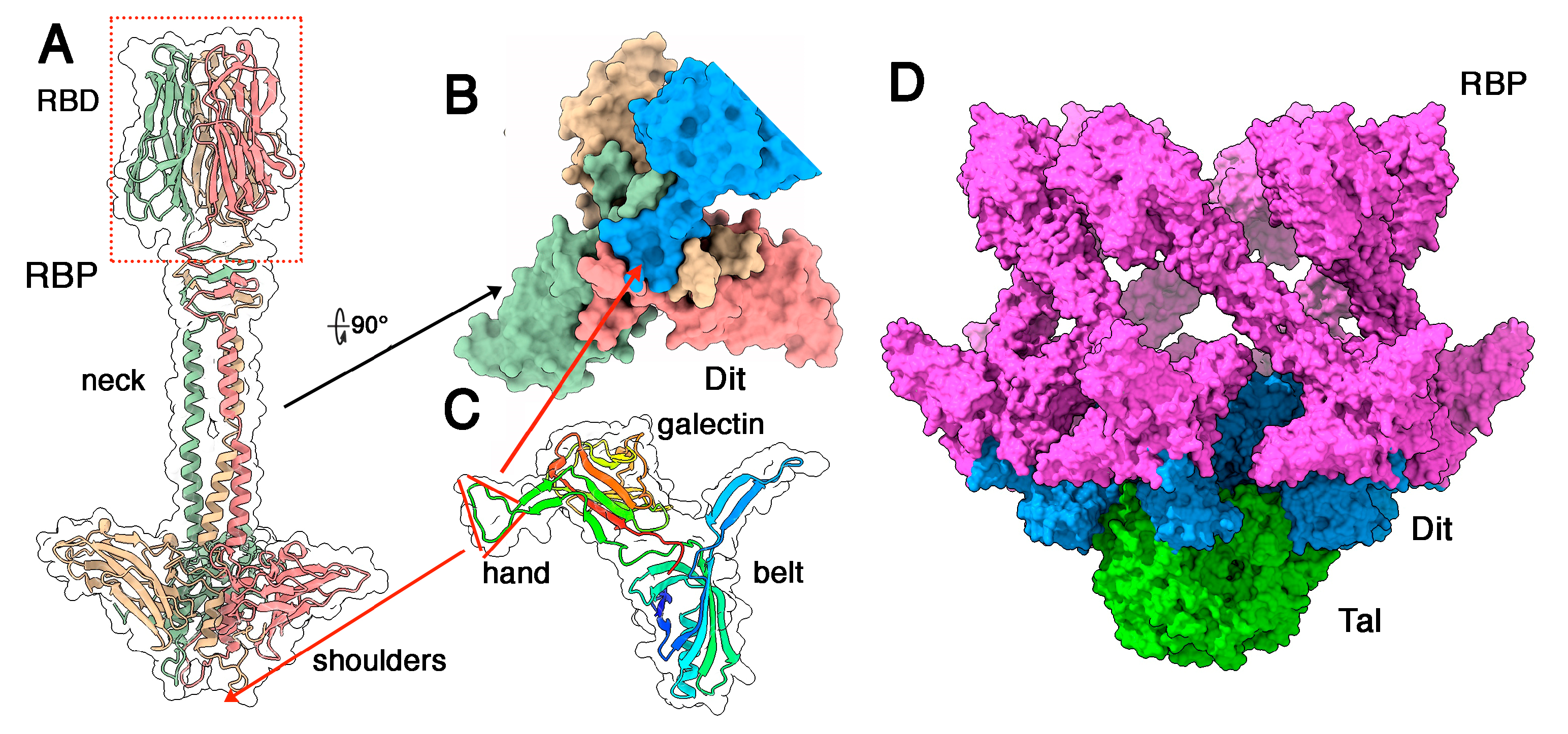
3.3. Phages Belonging to P335 Type III and IV: PhiLC3 and Q33
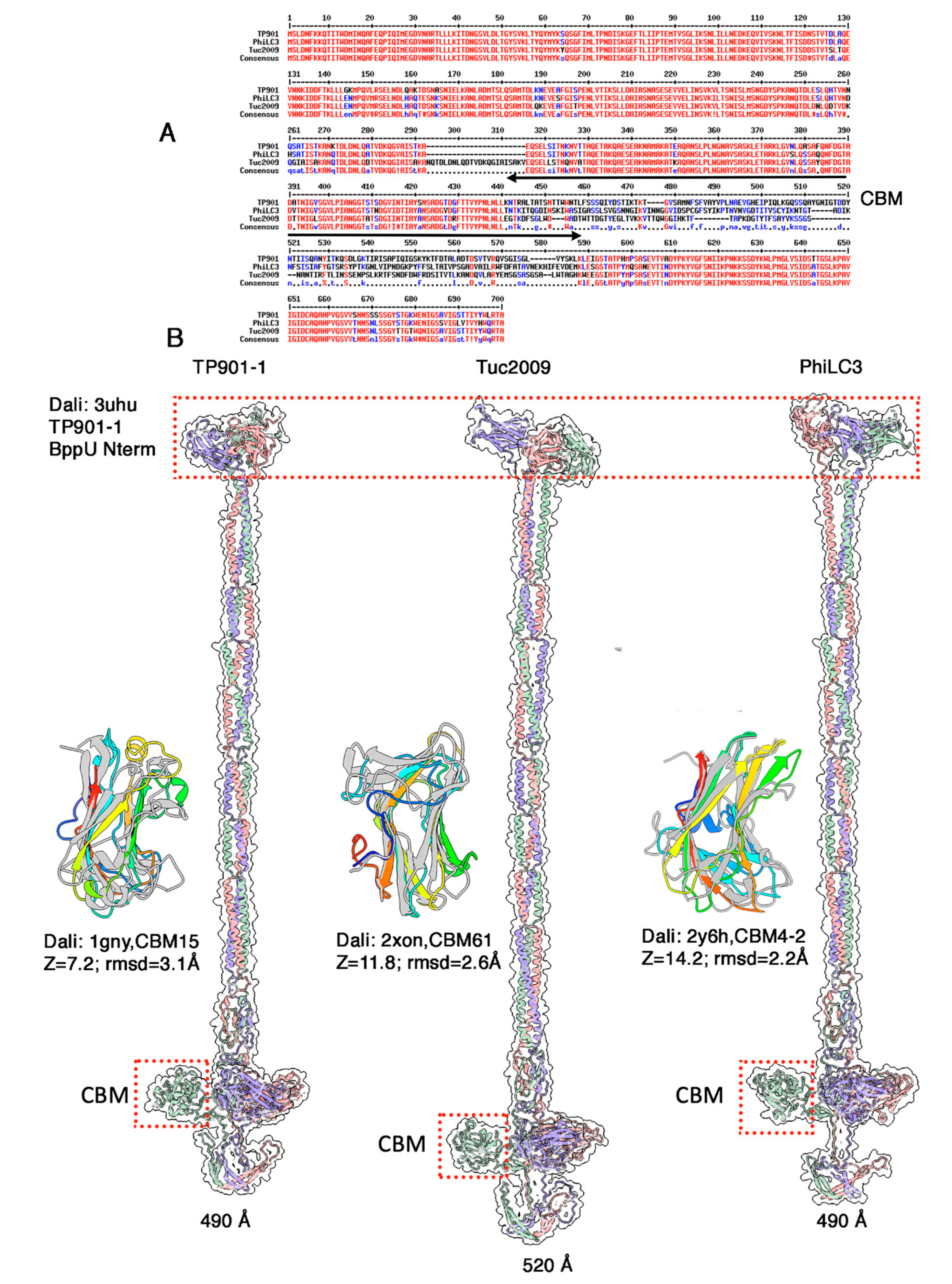
3.4. The Neck Passage Structure of Phages TP901-1, Tuc2009 and PhiLC3
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Labrie, S.J.; Josephsen, J.; Neve, H.; Vogensen, F.K.; Moineau, S. Morphology, genome sequence, and structural proteome of type phage P335 from Lactococcus lactis. Appl. Environ. Microbiol. 2008, 74, 4636–4644. [Google Scholar] [CrossRef] [Green Version]
- Mahony, J.; Oliveira, J.; Collins, B.; Hanemaaijer, L.; Lugli, G.A.; Neve, H.; Ventura, M.; Kouwen, T.R.; Cambillau, C.; van Sinderen, D. Genetic and functional characterisation of the lactococcal P335 phage-host interactions. BMC Genom. 2017, 18, 146. [Google Scholar] [CrossRef] [Green Version]
- Mahony, J.; Martel, B.; Tremblay, D.M.; Neve, H.; Heller, K.J.; Moineau, S.; van Sinderen, D. Identification of a new P335 subgroup through molecular analysis of lactococcal phages Q33 and BM13. Appl. Environ. Microbiol. 2013, 79, 4401–4409. [Google Scholar] [CrossRef] [Green Version]
- Mahanivong, C.; Boyce, J.D.; Davidson, B.E.; Hillier, A.J. Sequence analysis and molecular characterization of the Lactococcus lactis temperate bacteriophage BK5-T. Appl. Environ. Microbiol. 2001, 67, 3564–3576. [Google Scholar] [CrossRef] [Green Version]
- Blatny, J.M.; Godager, L.; Lunde, M.; Nes, I.F. Complete genome sequence of the Lactococcus lactis temperate phage phiLC3: Comparative analysis of phiLC3 and its relatives in lactococci and streptococci. Virology 2004, 318, 231–244. [Google Scholar] [CrossRef] [Green Version]
- Oliveira, J.; Mahony, J.; Hanemaaijer, L.; Kouwen, T.; van Sinderen, D. Biodiversity of bacteriophages infecting Lactococcus lactis starter cultures. J. Dairy Sci. 2018, 101, 96–105. [Google Scholar] [CrossRef]
- Boucher, I.; Emond, E.; Dion, E.; Montpetit, D.; Moineau, S. Microbiological and molecular impacts of AbiK on the lytic cycle of Lactococcus lactis phages of the 936 and P335 species. Microbiology 2000, 146, 445–453. [Google Scholar] [CrossRef] [Green Version]
- Kelly, W.J.; Altermann, E.; Lambie, S.C.; Leahy, S.C. Interaction between the genomes of Lactococcus lactis and phages of the P335 species. Front. Microbiol. 2013, 4, 257. [Google Scholar] [CrossRef] [Green Version]
- Collins, B.; Bebeacua, C.; Mahony, J.; Blangy, S.; Douillard, F.P.; Veesler, D.; Cambillau, C.; van Sinderen, D. Structure and functional analysis of the host recognition device of lactococcal phage tuc2009. J. Virol. 2013, 87, 8429–8440. [Google Scholar] [CrossRef] [Green Version]
- Shepherd, D.A.; Veesler, D.; Lichiere, J.; Ashcroft, A.E.; Cambillau, C. Unraveling lactococcal phage baseplate assembly by mass spectrometry. Mol. Cell Proteom. 2011, 10, M111.009787. [Google Scholar] [CrossRef]
- Legrand, P.; Collins, B.; Blangy, S.; Murphy, J.; Spinelli, S.; Gutierrez, C.; Richet, N.; Kellenberger, C.; Desmyter, A.; Mahony, J.; et al. The Atomic Structure of the Phage Tuc2009 Baseplate Tripod Suggests that Host Recognition Involves Two Different Carbohydrate Binding Modules. MBio 2016, 7, e01781-15. [Google Scholar] [CrossRef] [Green Version]
- Goulet, A.; Spinelli, S.; Mahony, J.; Cambillau, C. Conserved and Diverse Traits of Adhesion Devices from Siphoviridae Recognizing Proteinaceous or Saccharidic Receptors. Viruses 2020, 12, 512. [Google Scholar] [CrossRef]
- Philippe, C.; Levesque, S.; Dion, M.B.; Tremblay, D.M.; Horvath, P.; Luth, N.; Cambillau, C.; Franz, C.; Neve, H.; Fremaux, C.; et al. Novel Genus of Phages Infecting Streptococcus thermophilus: Genomic and Morphological Characterization. Appl. Environ. Microbiol. 2020, 86, e00227-20. [Google Scholar] [CrossRef]
- Kelleher, P.; Mahony, J.; Schweinlin, K.; Neve, H.; Franz, C.M.; van Sinderen, D. Assessing the functionality and genetic diversity of lactococcal prophages. Int. J. Food Microbiol. 2018, 272, 29–40. [Google Scholar] [CrossRef]
- Veesler, D.; Spinelli, S.; Mahony, J.; Lichiere, J.; Blangy, S.; Bricogne, G.; Legrand, P.; Ortiz-Lombardia, M.; Campanacci, V.; van Sinderen, D.; et al. Structure of the phage TP901-1 1.8 MDa baseplate suggests an alternative host adhesion mechanism. Proc. Natl. Acad. Sci. USA 2012, 109, 8954–8958. [Google Scholar] [CrossRef] [Green Version]
- Bebeacua, C.; Lai, L.; Vegge, C.S.; Brondsted, L.; van Heel, M.; Veesler, D.; Cambillau, C. Visualizing a complete Siphoviridae member by single-particle electron microscopy: The structure of lactococcal phage TP901-1. J. Virol. 2013, 87, 1061–1068. [Google Scholar] [CrossRef] [Green Version]
- Hayes, S.; Mahony, J.; Vincentelli, R.; Ramond, L.; Nauta, A.; van Sinderen, D.; Cambillau, C. Ubiquitous Carbohydrate Binding Modules Decorate 936 Lactococcal Siphophage Virions. Viruses 2019, 11, 631. [Google Scholar] [CrossRef] [Green Version]
- Hayes, S.; Vincentelli, R.; Mahony, J.; Nauta, A.; Ramond, L.; Lugli, G.A.; Ventura, M.; van Sinderen, D.; Cambillau, C. Functional carbohydrate binding modules identified in evolved dits from siphophages infecting various Gram-positive bacteria. Mol. Microbiol. 2018, 110, 777–795. [Google Scholar] [CrossRef]
- Lavelle, K.; Goulet, A.; McDonnell, B.; Spinelli, S.; van Sinderen, D.; Mahony, J.; Cambillau, C. Revisiting the host adhesion determinants of Streptococcus thermophilus siphophages. Microb. Biotechnol. 2020, 13, 1765–1779. [Google Scholar] [CrossRef]
- Goulet, A.; Joos, R.; Lavelle, K.; Van Sinderen, D.; Mahony, J.; Cambillau, C. A structural discovery journey of streptococcal phages adhesion devices by AlphaFold2. Front. Mol. Biosci. 2022, 9, 960325. [Google Scholar] [CrossRef]
- Mahony, J.; Frantzen, C.; Vinogradov, E.; Sadovskaya, I.; Theodorou, I.; Kelleher, P.; Chapot-Chartier, M.P.; Cambillau, C.; Holo, H.; van Sinderen, D. The CWPS Rubik’s cube: Linking diversity of cell wall polysaccharide structures with the encoded biosynthetic machinery of selected Lactococcus lactis strains. Mol. Microbiol. 2020, 114, 582–596. [Google Scholar] [CrossRef]
- Ostergaard Breum, S.; Neve, H.; Heller, K.J.; Vogensen, F.K. Temperate phages TP901-1 and phiLC3, belonging to the P335 species, apparently use different pathways for DNA injection in Lactococcus lactis subsp. cremoris 3107. FEMS Microbiol. Lett. 2007, 276, 156–164. [Google Scholar] [CrossRef] [Green Version]
- Veesler, D.; Robin, G.; Lichiere, J.; Auzat, I.; Tavares, P.; Bron, P.; Campanacci, V.; Cambillau, C. Crystal Structure of Bacteriophage SPP1 Distal Tail Protein (gp19.1): A BASEPLATE HUB PARADIGM IN GRAM-POSITIVE INFECTING PHAGES. J. Biol. Chem. 2010, 285, 36666–36673. [Google Scholar] [CrossRef] [Green Version]
- Flayhan, A.; Vellieux, F.M.; Lurz, R.; Maury, O.; Contreras-Martel, C.; Girard, E.; Boulanger, P.; Breyton, C. Crystal structure of pb9, the distal tail protein of bacteriophage T5: A conserved structural motif among all siphophages. J. Virol. 2014, 88, 820–828. [Google Scholar] [CrossRef] [Green Version]
- Goulet, A.; Cambillau, C. Structure and Topology Prediction of Phage Adhesion Devices Using AlphaFold2: The Case of Two Oenococcus oeni Phages. Microorganisms 2021, 9, 2151. [Google Scholar] [CrossRef]
- Veesler, D.; Cambillau, C. A common evolutionary origin for tailed-bacteriophage functional modules and bacterial machineries. Microbiol. Mol. Biol. Rev. MMBR 2011, 75, 423–433. [Google Scholar] [CrossRef] [Green Version]
- Spinelli, S.; Desmyter, A.; Verrips, C.T.; de Haard, H.J.; Moineau, S.; Cambillau, C. Lactococcal bacteriophage p2 receptor-binding protein structure suggests a common ancestor gene with bacterial and mammalian viruses. Nat. Struct. Mol. Biol. 2006, 13, 85–89. [Google Scholar] [CrossRef]
- Sciara, G.; Bebeacua, C.; Bron, P.; Tremblay, D.; Ortiz-Lombardia, M.; Lichiere, J.; van Heel, M.; Campanacci, V.; Moineau, S.; Cambillau, C. Structure of lactococcal phage p2 baseplate and its mechanism of activation. Proc. Natl. Acad. Sci. USA 2010, 107, 6852–6857. [Google Scholar] [CrossRef] [Green Version]
- Kizziah, J.L.; Manning, K.A.; Dearborn, A.D.; Dokland, T. Structure of the host cell recognition and penetration machinery of a Staphylococcus aureus bacteriophage. PLOS Pathog. 2020, 12, e1008314. [Google Scholar]
- Dunne, M.; Rupf, B.; Tala, M.; Qabrati, X.; Ernst, P.; Shen, Y.; Sumrall, E.; Heeb, L.; Pluckthun, A.; Loessner, M.J.; et al. Reprogramming Bacteriophage Host Range through Structure-Guided Design of Chimeric Receptor Binding Proteins. Cell Rep. 2019, 29, 1336–1350.e4. [Google Scholar] [CrossRef] [Green Version]
- Dieterle, M.E.; Fina Martin, J.; Duran, R.; Nemirovsky, S.I.; Sanchez Rivas, C.; Bowman, C.; Russell, D.; Hatfull, G.F.; Cambillau, C.; Piuri, M. Characterization of prophages containing “evolved” Dit/Tal modules in the genome of Lactobacillus casei BL23. Appl. Microbiol. Biotechnol. 2016, 100, 9201–9215. [Google Scholar] [CrossRef]
- Dieterle, M.E.; Spinelli, S.; Sadovskaya, I.; Piuri, M.; Cambillau, C. Evolved distal tail carbohydrate binding modules of Lactobacillus phage J-1: A novel type of anti-receptor widespread among lactic acid bacteria phages. Mol. Microbiol. 2017, 104, 608–620. [Google Scholar] [CrossRef] [Green Version]
- Stockdale, S.R.; Mahony, J.; Courtin, P.; Chapot-Chartier, M.P.; van Pijkeren, J.P.; Britton, R.A.; Neve, H.; Heller, K.J.; Aideh, B.; Vogensen, F.K.; et al. The lactococcal phages Tuc2009 and TP901-1 incorporate two alternate forms of their tail fiber into their virions for infection specialization. J. Biol. Chem. 2013, 288, 5581–5590. [Google Scholar] [CrossRef] [Green Version]
- Sao-Jose, C.; Lhuillier, S.; Lurz, R.; Melki, R.; Lepault, J.; Santos, M.A.; Tavares, P. The ectodomain of the viral receptor YueB forms a fiber that triggers ejection of bacteriophage SPP1 DNA. J. Biol. Chem. 2006, 281, 11464–11470. [Google Scholar] [CrossRef] [Green Version]
- Linares, R.; Arnaud, C.A.; Degroux, S.; Schoehn, G.; Breyton, C. Structure, function and assembly of the long, flexible tail of siphophages. Curr. Opin. Virol. 2020, 45, 34–42. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Zidek, A.; Potapenko, A.; et al. Applying and improving AlphaFold at CASP14. Proteins 2021, 89, 1711–1721. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Zidek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Evans, R.; O’Neill, M.; Pritzel, A.; Antropova, N.; Senior, A.; Green, T.; Žídek, A.; Bates, R.; Blackwell, S.; Yim, J.; et al. Protein complex prediction with AlphaFold-Multimer. BioRxiv 2021. [Google Scholar] [CrossRef]
- Huang, Y.J.; Zhang, N.; Bersch, B.; Fidelis, K.; Inouye, M.; Ishida, Y.; Kryshtafovych, A.; Kobayashi, N.; Kuroda, Y.; Liu, G.; et al. Assessment of prediction methods for protein structures determined by NMR in CASP14: Impact of AlphaFold2. Proteins 2021, 89, 1959–1976. [Google Scholar] [CrossRef]
- Goulet, A.; Cambillau, C. Present impact of AlphaFold2 revolution on structural biology, and an illustration with the structure prediction of the bacteriophage J-1 host adhesion device. Front. Mol. Biosci. 2022, in press. [Google Scholar]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 486–501. [Google Scholar] [CrossRef] [Green Version]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Meng, E.C.; Couch, G.S.; Croll, T.I.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Structure visualization for researchers, educators, and developers. Protein Sci. 2021, 30, 70–82. [Google Scholar] [CrossRef]
- Holm, L. DALI and the persistence of protein shape. Protein Sci. 2020, 29, 128–140. [Google Scholar] [CrossRef] [Green Version]
- Corpet, F. Multiple sequence alignment with hierarchical clustering. Nucleic Acids Res. 1988, 16, 10881–10890. [Google Scholar] [CrossRef]
- Gouet, P.; Robert, X.; Courcelle, E. ESPript/ENDscript: Extracting and rendering sequence and 3D information from atomic structures of proteins. Nucleic Acids Res. 2003, 31, 3320–3323. [Google Scholar] [CrossRef]
- Krissinel, E.; Henrick, K. Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 2007, 372, 774–797. [Google Scholar] [CrossRef]
- Ricagno, S.; Campanacci, V.; Blangy, S.; Spinelli, S.; Tremblay, D.; Moineau, S.; Tegoni, M.; Cambillau, C. Crystal structure of the receptor-binding protein head domain from Lactococcus lactis phage bIL170. J. Virol. 2006, 80, 9331–9335. [Google Scholar] [CrossRef] [Green Version]
- Fujimoto, Z.; Suzuki, N.; Kishine, N.; Ichinose, H.; Momma, M.; Kimura, A.; Funane, K. Carbohydrate-binding architecture of the multi-modular alpha-1,6-glucosyltransferase from Paenibacillus sp. 598K, which produces alpha-1,6-glucosyl-alpha-glucosaccharides from starch. Biochem. J. 2017, 474, 2763–2778. [Google Scholar] [CrossRef]
- Suits, M.D.L.; Pluvinage, B.; Law, A.; Liu, Y.; Palma, A.S.; Chai, W.; Feizi, T.; Boraston, A.B. Conformational analysis of the Streptococcus pneumoniae hyaluronate lyase and characterization of its hyaluronan-specific carbohydrate-binding module. J. Biol. Chem. 2014, 289, 27264–27277. [Google Scholar] [CrossRef] [Green Version]
- Spinelli, S.; Campanacci, V.; Blangy, S.; Moineau, S.; Tegoni, M.; Cambillau, C. Modular structure of the receptor binding proteins of Lactococcus lactis phages. The RBP structure of the temperate phage TP901-1. J. Biol. Chem. 2006, 281, 14256–14262. [Google Scholar] [CrossRef] [Green Version]
- Campanacci, V.; Veesler, D.; Lichiere, J.; Blangy, S.; Sciara, G.; Moineau, S.; van Sinderen, D.; Bron, P.; Cambillau, C. Solution and electron microscopy characterization of lactococcal phage baseplates expressed in Escherichia coli. J. Struct. Biol. 2010, 172, 75–84. [Google Scholar] [CrossRef]
- Kanamaru, S.; Leiman, P.G.; Kostyuchenko, V.A.; Chipman, P.R.; Mesyanzhinov, V.V.; Arisaka, F.; Rossmann, M.G. Structure of the cell-puncturing device of bacteriophage T4. Nature 2002, 415, 553–557. [Google Scholar] [CrossRef]
- Xiang, Y.; Morais, M.C.; Cohen, D.N.; Bowman, V.D.; Anderson, D.L.; Rossmann, M.G. Crystal and cryoEM structural studies of a cell wall degrading enzyme in the bacteriophage phi29 tail. Proc. Natl. Acad. Sci. USA 2008, 105, 9552–9557. [Google Scholar] [CrossRef] [Green Version]
- Firczuk, M.; Mucha, A.; Bochtler, M. Crystal structures of active LytM. J. Mol. Biol. 2005, 354, 578–590. [Google Scholar] [CrossRef]
- Vegge, C.S.; Neve, H.; Brondsted, L.; Heller, K.J.; Vogensen, F.K. Analysis of the collar-whisker structure of temperate lactococcal bacteriophage TP901-1. Appl. Environ. Microbiol. 2006, 72, 6815–6818. [Google Scholar] [CrossRef] [Green Version]
- Kenny, J.G.; McGrath, S.; Fitzgerald, G.F.; van Sinderen, D. Bacteriophage Tuc2009 encodes a tail-associated cell wall-degrading activity. J. Bacteriol. 2004, 186, 3480–3491. [Google Scholar] [CrossRef] [Green Version]
- Cid, M.; Pedersen, H.L.; Kaneko, S.; Coutinho, P.M.; Henrissat, B.; Willats, W.G.; Boraston, A.B. Recognition of the helical structure of beta-1,4-galactan by a new family of carbohydrate-binding modules. J. Biol. Chem 2010, 285, 35999–36009. [Google Scholar] [CrossRef] [Green Version]
- Von Schantz, L.; Hakansson, M.; Logan, D.T.; Walse, B.; Osterlin, J.; Nordberg-Karlsson, E.; Ohlin, M. Structural basis for carbohydrate-binding specificity--a comparative assessment of two engineered carbohydrate-binding modules. Glycobiology 2012, 22, 948–961. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Goulet, A.; Mahony, J.; Cambillau, C.; van Sinderen, D. Exploring Structural Diversity among Adhesion Devices Encoded by Lactococcal P335 Phages with AlphaFold2. Microorganisms 2022, 10, 2278. https://doi.org/10.3390/microorganisms10112278
Goulet A, Mahony J, Cambillau C, van Sinderen D. Exploring Structural Diversity among Adhesion Devices Encoded by Lactococcal P335 Phages with AlphaFold2. Microorganisms. 2022; 10(11):2278. https://doi.org/10.3390/microorganisms10112278
Chicago/Turabian StyleGoulet, Adeline, Jennifer Mahony, Christian Cambillau, and Douwe van Sinderen. 2022. "Exploring Structural Diversity among Adhesion Devices Encoded by Lactococcal P335 Phages with AlphaFold2" Microorganisms 10, no. 11: 2278. https://doi.org/10.3390/microorganisms10112278