Complex Cell Type-Specific Roles of Autophagy in Liver Fibrosis and Cirrhosis


Abstract
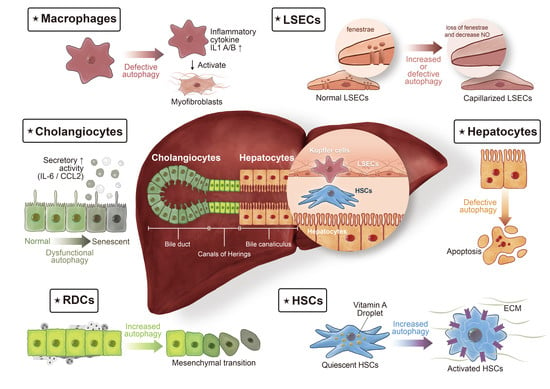
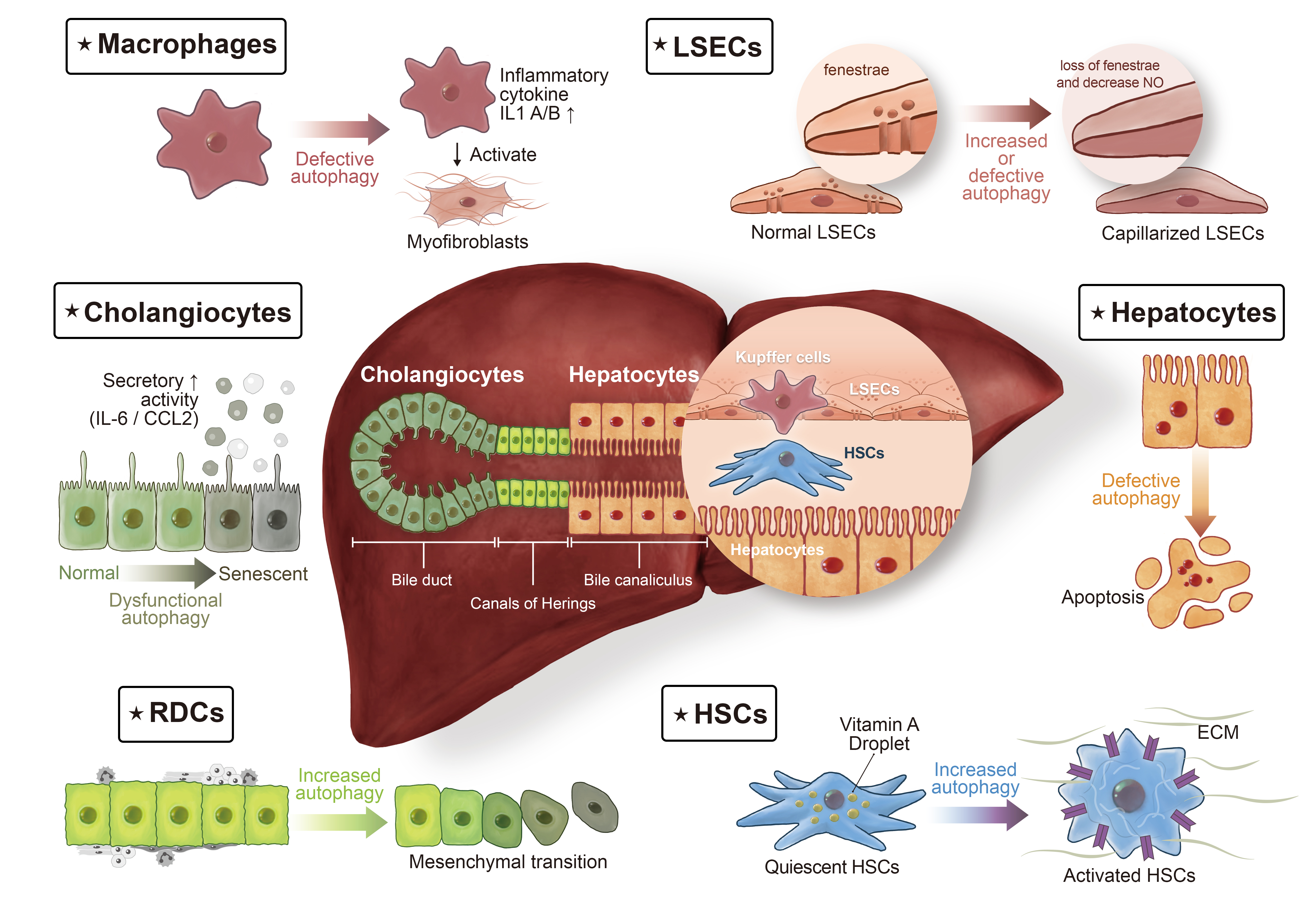
:
{kind=link}
{kind=link}
1. Introduction
2. Autophagy
3. Cellular and Molecular Determinants of Liver Fibrosis
3.1. HSCs
3.2. Reactive Ductular Cells
3.3. Hepatocytes
3.4. Cholangiocytes
3.5. Macrophages
3.6. Liver Sinusoidal Endothelial Cells
4. Conclusions and Future Perspectives
4.1. Challenges in the Development of Autophagy Modulators
4.2. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Teckman, J.H.; Perlmutter, D.H. Retention of mutant alpha (1)-antitrypsin Z in endoplasmic reticulum is associated with an autophagic response. Am. J. Physiol. Gastrointest. Liver Physiol. 2000, 279, G961–G974. [Google Scholar] [CrossRef] [PubMed]
- Kamimoto, T.; Shoji, S.; Hidvegi, T.; Mizushima, N.; Umebayashi, K.; Perlmutter, D.H.; Yoshimori, T. Intracellular inclusions containing mutant alpha1-antitrypsin Z are propagated in the absence of autophagic activity. J. Biol. Chem. 2006, 281, 4467–4476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hidvegi, T.; Ewing, M.; Hale, P.; Dippold, C.; Beckett, C.; Kemp, C.; Maurice, N.; Mukherjee, A.; Goldbach, C.; Watkins, S.; et al. An autophagy-enhancing drug promotes degradation of mutant alpha1-antitrypsin Z and reduces hepatic fibrosis. Science 2010, 329, 229–232. [Google Scholar] [CrossRef]
- Ding, W.X.; Li, M.; Chen, X.; Ni, H.M.; Lin, C.W.; Gao, W.; Lu, B.; Stolz, D.B.; Clemens, D.L.; Yin, X.M. Autophagy reduces acute ethanol-induced hepatotoxicity and steatosis in mice. Gastroenterology 2010, 139, 1740–1752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, C.W.; Zhang, H.; Li, M.; Xiong, X.; Chen, X.; Chen, X.; Dong, X.C.; Yin, X.M. Pharmacological promotion of autophagy alleviates steatosis and injury in alcoholic and non-alcoholic fatty liver conditions in mice. J. Hepatol. 2013, 58, 993–999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukuo, Y.; Yamashina, S.; Sonoue, H.; Arakawa, A.; Nakadera, E.; Aoyama, T.; Uchiyama, A.; Kon, K.; Ikejima, K.; Watanabe, S. Abnormality of autophagic function and cathepsin expression in the liver from patients with non-alcoholic fatty liver disease. Hepatol. Res. 2014, 44, 1026–1036. [Google Scholar] [CrossRef]
- Sir, D.; Tian, Y.; Chen, W.L.; Ann, D.K.; Yen, T.S.; Ou, J.H. The early autophagic pathway is activated by hepatitis B virus and required for viral DNA replication. Proc. Natl Acad. Sci. USA 2010, 107, 4383–4388. [Google Scholar] [CrossRef] [Green Version]
- Qu, X.; Yu, J.; Bhagat, G.; Furuya, N.; Hibshoosh, H.; Troxel, A.; Rosen, J.; Eskelinen, E.L.; Mizushima, N.; Ohsumi, Y.; et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J. Clin. Investig. 2003, 112, 1809–1820. [Google Scholar] [CrossRef] [Green Version]
- Sir, D.; Chen, W.L.; Choi, J.; Wakita, T.; Yen, T.S.; Ou, J.H. Induction of incomplete autophagic response by hepatitis C virus via the unfolded protein response. Hepatology 2008, 48, 1054–1061. [Google Scholar] [CrossRef] [Green Version]
- Rautou, P.E.; Cazals-Hatem, D.; Feldmann, G.; Mansouri, A.; Grodet, A.; Barge, S.; Martinot-Peignoux, M.; Duces, A.; Bièche, I.; Lebrec, D.; et al. Changes in autophagic response in patients with chronic hepatitis C virus infection. Am. J. Pathol. 2011, 178, 2708–2715. [Google Scholar] [CrossRef] [Green Version]
- Takamura, A.; Komatsu, M.; Hara1, T.; Sakamoto, A.; Kishi, C.; Waguri, S.; Eishi, Y.; Hino, O.; Tanaka, K.; Mizushima, N. Autophagy-deficient mice develop multiple liver tumors. Genes Dev. 2011, 25, 795–800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, D.H.; Jia, C.C.; Chen, J.; Lin, Z.X.; Ruan, D.Y.; Li, X.; Lin, Q.; Dong, M.; Ma, X.K.; Wan, X.B.; et al. Autophagic LC3B overexpression correlates with malignant progression and predicts a poor prognosis in hepatocellular carcinoma. Tumor Biol. 2014, 35, 12225–12233. [Google Scholar] [CrossRef] [PubMed]
- Gracia-Sancho, J.; Guixé-Muntet, S. The many-faced role of autophagy in liver diseases. J. Hepatol. 2018, 68, 593–594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allaire, M.; Rautou, P.E.; Codogno, P.; Lotersztajn, S. Autophagy in liver diseases: Time for translation? J. Hepatol. 2019, 70, 985–998. [Google Scholar] [CrossRef] [Green Version]
- Weiskirchen, R.; Tacke, F. Relevance of autophagy in parenchymal and non-parenchymal liver cells for health and disease. Cells 2019, 8, 16. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; Klionsky, D.J. Eaten alive: A history of macroautophagy. Nat. Cell Biol. 2010, 12, 814–822. [Google Scholar] [CrossRef] [Green Version]
- Mizushima, N.; Yoshimori, T.; Levine, B. Methods in mammalian autophagy research. Cell 2010, 140, 313–326. [Google Scholar] [CrossRef] [Green Version]
- Mizushima, N.; Yoshimori, T.; Ohsumi, Y. The role of Atg proteins in autophagosome formation. Annu. Rev. Cell Dev. Biol. 2011, 27, 107–132. [Google Scholar] [CrossRef]
- Kabeya, Y.; Mizushima, N.; Yamamoto, A.; Oshitani-Okamoto, S.; Ohsumi, Y.; Yoshimori, T. LC3, GABARAP and GATE16 localize to autophagosomal membrane depending on form-II formation. J. Cell Sci. 2004, 117, 2805–2812. [Google Scholar] [CrossRef] [Green Version]
- Kabeya, Y.; Mizushima, N.; Ueno, T.; Yamamoto, A.; Kirisako, T.; Noda, T.; Kominami, E.; Ohsumi, Y.; Yoshimori, T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000, 19, 5720–5728. [Google Scholar] [CrossRef]
- Bjørkøy, G.; Lamark, T.; Brech, A.; Outzen, H.; Perander, M.; Øvervatn, A.; Stenmark, H.; Johansen, T. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J. Cell Biol. 2005, 171, 603–614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizushima, N.; Yoshimori, T. How to interpret LC3 immunoblotting. Autophagy 2007, 3, 542–545. [Google Scholar] [CrossRef] [PubMed]
- Deter, R.L.; Baudhuin, P.; De Duve, C. Participation of lysosomes in cellular autophagy induced in rat liver by glucagon. J. Cell Biol. 1967, 35, C11–C16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madrigal-Matute, J.; Cuervo, A.M. Regulation of liver metabolism by autophagy. Gastroenterology 2016, 150, 328–339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Czaja, M.J.; Ding, W.X.; Donohue, T.M., Jr.; Friedman, S.L.; Kim, J.S.; Komatsu, M.; Lemasters, J.J.; Lemoine, A.; Lin, J.D.; Ou, J.H.; et al. Functions of autophagy in normal and diseased liver. Autophagy 2013, 9, 1131–1158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedman, S.L. Mechanisms of hepatic fibrogenesis. Gastroenterology 2008, 134, 1655–1669. [Google Scholar] [CrossRef] [Green Version]
- Tsuchida, T.; Friedman, S.L. Mechanisms of hepatic stellate cell activation. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 397–411. [Google Scholar] [CrossRef]
- Williams, M.J.; Clouston, A.D.; Forbes, S.J. Links between hepatic fibrosis, ductular reaction, and progenitor cell expansion. Gastroenterology 2014, 146, 349–356. [Google Scholar] [CrossRef]
- Sato, K.; Marzioni, M.; Meng, F.; Francis, H.; Glaser, S.; Alpini, G. Ductular reaction in liver diseases: Pathological mechanisms and translational significances. Hepatology 2019, 69, 420–430. [Google Scholar] [CrossRef] [Green Version]
- Gouw, A.H.; Clouston, A.D.; Theise, N.D. Ductular reactions in human liver: Diversity at the interface. Hepatology 2011, 54, 1853–1863. [Google Scholar] [CrossRef]
- Clouston, A.D.; Powell, E.E.; Walsh, M.J.; Richardson, M.M.; Demetris, A.J.; Jonsson, J.R. Fibrosis correlates with a ductular reaction in hepatitis C: Roles of impaired replication, progenitor cells and steatosis. Hepatology 2005, 41, 809–818. [Google Scholar] [CrossRef] [PubMed]
- Richardson, M.M.; Jonsson, J.R.; Powell, E.E.; Brunt, E.M.; Neuschwander-Tetri, B.A.; Bhathal, P.S.; Dixon, J.B.; Weltman, M.D.; Tilg, H.; Moschen, A.R.; et al. Progressive fibrosis in nonalcoholic steatohepatitis: Association with altered regeneration and a ductular reaction. Gastroenterology 2007, 133, 80–90. [Google Scholar] [CrossRef] [PubMed]
- Wood, M.J.; Gadd, V.L.; Powell, L.W.; Ramm, G.A.; Clouston, A.D. Ductular reaction in hereditary hemochromatosis: The link between hepatocyte senescence and fibrosis progression. Hepatology 2014, 59, 848–857. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Gea, V.; Ghiassi-Nejad, Z.; Rozenfeld, R.; Gordon, R.; Fiel, M.I.; Yue, Z.; Czaja, M.J.; Friedman, S.L. Autophagy releases lipid that promotes fibrogenesis by activated hepatic stellate cells in mice and in human tissues. Gastroenterology 2012, 142, 938–946. [Google Scholar] [CrossRef] [Green Version]
- Thoen, L.F.; Guimarães, E.L.; Dollé, L.; Mannaerts, I.; Najimi, M.; Sokal, E.; van Grunsven, L.A. A role for autophagy during hepatic stellate cell activation. J. Hepatol. 2011, 55, 1353–1360. [Google Scholar] [CrossRef]
- Hung, T.M.; Yuan, R.H.; Huang, W.P.; Chen, Y.H.; Lin, Y.C.; Lin, C.W.; Lai, H.S.; Lee, P.H. Increased autophagy markers are associated with ductular reaction during the development of cirrhosis. Am. J. Pathol. 2015, 185, 2454–2467. [Google Scholar] [CrossRef]
- Pozniak, K.N.; Pearen, M.A.; Pereira, T.N.; Kramer, C.S.M.; Kalita-De Croft, P.; Nawaratna, S.K.; Fernandez-Rojo, M.A.; Gobert, G.N.; Tirnitz-Parker, J.E.E.; Olynyk, J.K.; et al. Taurocholate induces biliary differentiation of liver progenitor cells causing hepatic stellate cell chemotaxis in the ductular reaction: Role in pediatric cystic fibrosis liver disease. Am. J. Pathol. 2017, 187, 2744–2757. [Google Scholar] [CrossRef] [Green Version]
- Hung, T.M.; Huang, Y.J.; Lin, Y.C.; Chen, Y.H.; Wu, Y.M.; Lee, P.H. A critical role of autophagy in regulating the mesenchymal transition of ductular cells in liver cirrhosis. Sci. Rep. 2019, 9, 10673. [Google Scholar] [CrossRef]
- Feldstein, A.E.; Canbay, A.; Angulo, P.; Taniai, M.; Burgart, L.J.; Lindor, K.D.; Gores, G.J. Hepatocyte apoptosis and fas expression are prominent features of human nonalcoholic steatohepatitis. Gastroenterology 2003, 125, 437–443. [Google Scholar] [CrossRef]
- Ni, H.M.; Bockus, A.; Boggess, N.; Jaeschke, H.; Ding, W.X. Activation of autophagy protects against acetaminophen-induced hepatotoxicity. Hepatology 2012, 55, 222–232. [Google Scholar] [CrossRef] [Green Version]
- Amir, M.; Zhao, E.; Fontana, L.; Rosenberg, H.; Tanaka, K.; Gao, G.; Czaja, M.J. Inhibition of hepatocyte autophagy increases tumor necrosis factor-dependent liver injury by promoting caspase-8 activation. Cell Death Differ. 2013, 20, 878–887. [Google Scholar] [CrossRef] [PubMed]
- Ni, H.M.; Woolbright, B.L.; Williams, J.; Copple, B.; Cui, W.; Luyendyk, J.P.; Jaeschke, H.; Ding, W.X. Nrf2 promotes the development of fibrosis and tumorigenesis in mice with defective hepatic autophagy. J. Hepatol. 2014, 61, 617–625. [Google Scholar] [CrossRef] [Green Version]
- Schläfli, A.M.; Berezowska, S.; Adams, O.; Langer, R.; Tschan, M.P. Reliable LC3 and p62 autophagy marker detection in formalin fixed paraffin embedded human tissue by immunohistochemistry. Eur. J. Histochem. 2015, 59, 173–179. [Google Scholar] [CrossRef] [Green Version]
- Nakanuma, Y.; Sasaki, M.; Harada, K. Autophagy and senescence in fibrosing cholangiopathies. J. Hepatol. 2015, 62, 934–945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sasaki, M.; Miyakoshi, M.; Sato, Y.; Nakanuma, Y. Autophagy mediates the process of cellular senescence characterizing bile duct damages in primary biliary cirrhosis. Lab Investig. 2010, 90, 835–843. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, M.; Miyakoshi, M.; Sato, Y.; Nakanuma, Y. A possible involvement of p62/sequestosome-1 in the process of biliary epithelial autophagy and senescence in primary biliary cirrhosis. Liver Int. 2012, 32, 487–499. [Google Scholar] [CrossRef] [PubMed]
- Krenkel, O.; Tacke, F. Liver macrophages in tissue homeostasis and disease. Nat. Rev. Immunol. 2017, 17, 306–321. [Google Scholar] [CrossRef]
- Ding, W.X.; Jaeschke, H. Autophagy in macrophages regulates the inflammasome and protects against liver injury. J. Hepatol. 2016, 64, 16–18. [Google Scholar] [CrossRef] [Green Version]
- Liu, K.; Zhao, E.; Ilyas, G.; Lalazar, G.; Lin, Y.; Haseeb, M.; Tanaka, K.E.; Czaja, M.J. Impaired macrophage autophagy increases the immune response in obese mice by promoting proinflammatory macrophage polarization. Autophagy 2015, 11, 271–284. [Google Scholar] [CrossRef] [Green Version]
- Lodder, J.; Denaës, T.; Chobert, M.N.; Wan, J.; El-Benna, J.; Pawlotsky, J.M.; Lotersztajn, S.; Teixeira-Clerc, F. Macrophage autophagy protects against liver fibrosis in mice. Autophagy 2015, 11, 1280–1292. [Google Scholar] [CrossRef] [Green Version]
- Poisson, J.; Lemoinne, S.; Boulanger, C.; Durand, F.; Moreau, R.; Valla, D.; Rautou, P.E. Liver sinusoidal endothelial cells: Physiology and role in liver diseases. J. Hepatol. 2017, 66, 212–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, X.; Wang, D.; Zhu, X.; Wang, G.; You, Y.; Ning, Z.; Li, Y.; Jin, S.; Huang, Y.; Hu, Y.; et al. Autophagic degradation of caveolin-1 promotes liver sinusoidal endothelial cells defenestration. Cell Death Dis. 2018, 9, 576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruart, M.; Chavarria, L.; Campreciós, G.; Suárez-Herrera, N.; Montironi, C.; Guixé-Muntet, S.; Bosch, J.; Friedman, S.L.; Garcia-Pagán, J.C.; Hernández-Gea, V. Impaired endothelial autophagy promotes liver fibrosis by aggravating the oxidative stress response during acute liver injury. J. Hepatol. 2019, 70, 458–469. [Google Scholar] [CrossRef] [PubMed]
- Gracia-Sancho, J.; Guixé-Muntet, S.; Hide, D.; Bosch, J. Modulation of autophagy for the treatment of liver diseases. Expert Opin. Investig. Drugs 2014, 23, 965–977. [Google Scholar] [CrossRef]
- Del Principe, D.; Lista, P.; Malomi, W.; Giammarioli, A.M. Fibroblast autophagy in fibrotic disorder. J. Pathol. 2013, 229, 208–220. [Google Scholar] [CrossRef]
- Schuppana, D.; Pinzani, M. Anti-fibrotic therapy: Lost in translation? J. Hepatol. 2012, 56, S66–S74. [Google Scholar] [CrossRef]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hung, T.-M.; Hsiao, C.-C.; Lin, C.-W.; Lee, P.-H. Complex Cell Type-Specific Roles of Autophagy in Liver Fibrosis and Cirrhosis. Pathogens 2020, 9, 225. https://doi.org/10.3390/pathogens9030225
Hung T-M, Hsiao C-C, Lin C-W, Lee P-H. Complex Cell Type-Specific Roles of Autophagy in Liver Fibrosis and Cirrhosis. Pathogens. 2020; 9(3):225. https://doi.org/10.3390/pathogens9030225
Chicago/Turabian StyleHung, Tzu-Min, Chih-Chiang Hsiao, Chih-Wen Lin, and Po-Huang Lee. 2020. "Complex Cell Type-Specific Roles of Autophagy in Liver Fibrosis and Cirrhosis" Pathogens 9, no. 3: 225. https://doi.org/10.3390/pathogens9030225