
Analysis of CRISPR/Cas Genetic Structure, Spacer Content and Molecular Epidemiology in Brazilian Acinetobacter baumannii Clinical Isolates

 and
and
Abstract
:1. Introduction
2. Results
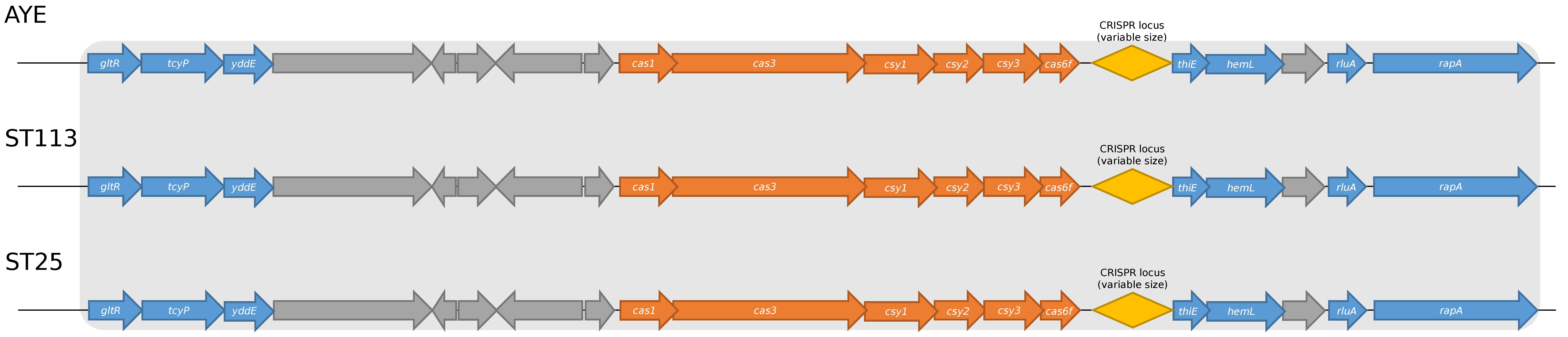
2.1. CRISPR/Cas Genetic Structure in Brazilian A. baumannii Isolates
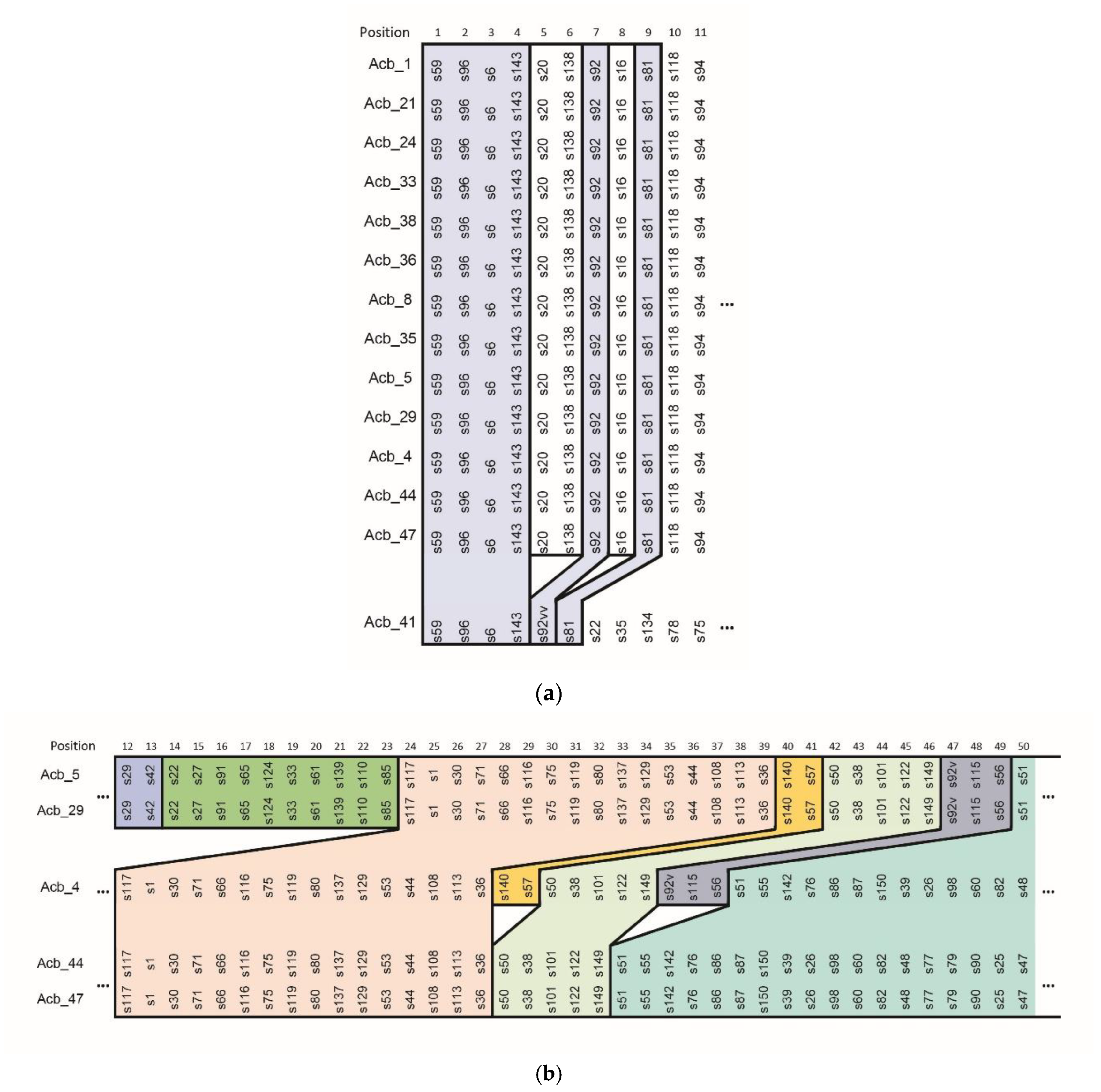
2.2. Spacer Analysis
2.3. Prophage Identification
2.4. CRISPR Typing
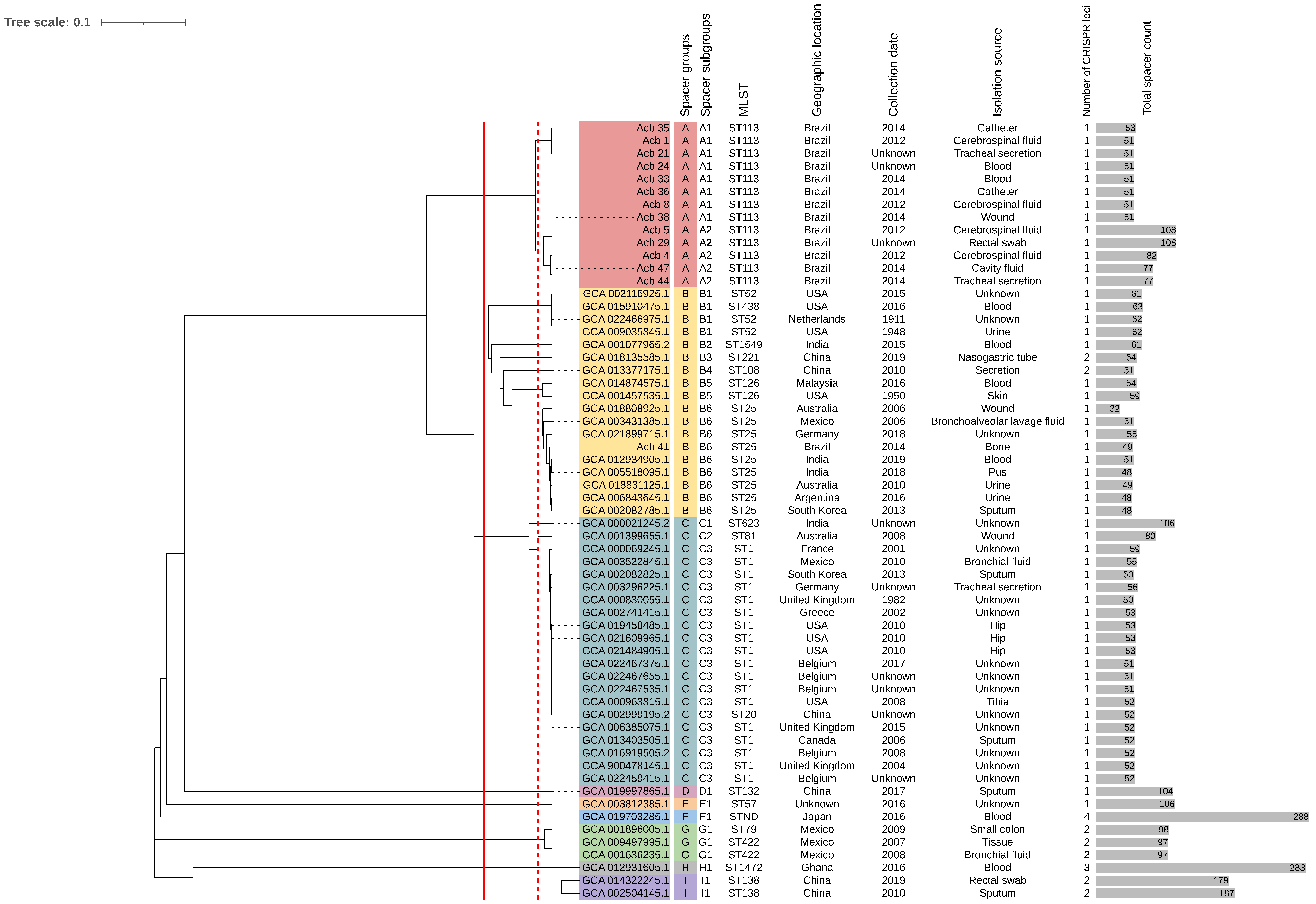
2.5. Phylogenetic and Epidemiologic Approaches
3. Discussion
4. Materials and Methods
4.1. Brazilian Bacterial Strains
4.2. CRISPR/Cas Analysis
4.3. Anti-CRISPR Investigation
4.4. Leader Sequence
4.5. Spacer Analysis
4.6. Bacteriophage Investigation
4.7. Phylogeny and Epidemiology Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Frost, L.S.; Leplae, R.; Summers, A.O.; Toussaint, A. Mobile Genetic Elements: The Agents of Open Source Evolution. Nat. Rev. Microbiol. 2005, 3, 722–732. [Google Scholar] [CrossRef]
- Labrie, S.J.; Samson, J.E.; Moineau, S. Bacteriophage Resistance Mechanisms. Nat. Rev. Microbiol. 2010, 8, 317–327. [Google Scholar] [CrossRef]
- Barrangou, R.; Fremaux, C.; Deveau, H.; Richards, M.; Boyaval, P.; Moineau, S.; Romero, D.A.; Horvath, P. CRISPR Provides Acquired Resistance Against Viruses in Prokaryotes. Science 2007, 315, 1709–1712. [Google Scholar] [CrossRef] [PubMed]
- Makarova, K.S.; Wolf, Y.I.; Alkhnbashi, O.S.; Costa, F.; Shah, S.A.; Saunders, S.J.; Barrangou, R.; Brouns, S.J.J.; Charpentier, E.; Haft, D.H.; et al. An Updated Evolutionary Classification of CRISPR–Cas Systems. Nat. Rev. Microbiol. 2015, 13, 722–736. [Google Scholar] [CrossRef]
- Jansen, R.; Embden, J.D.V.; Gaastra, W.; Schouls, L.M. Identification of Genes That Are Associated with DNA Repeats in Prokaryotes. Mol. Microbiol. 2002, 43, 1565–1575. [Google Scholar] [CrossRef]
- Haft, D.H.; Selengut, J.; Mongodin, E.F.; Nelson, K.E. A Guild of 45 CRISPR-Associated (Cas) Protein Families and Multiple CRISPR/Cas Subtypes Exist in Prokaryotic Genomes. PLoS Comput. Biol. 2005, 1, e60. [Google Scholar] [CrossRef]
- Koonin, E.V.; Makarova, K.S.; Zhang, F. Diversity, Classification and Evolution of CRISPR-Cas Systems. Curr. Opin. Microbiol. 2017, 37, 67–78. [Google Scholar] [CrossRef]
- Bondy-Denomy, J.; Pawluk, A.; Maxwell, K.L.; Davidson, A.R. Bacteriophage Genes That Inactivate the CRISPR/Cas Bacterial Immune System. Nature 2013, 493, 429–432. [Google Scholar] [CrossRef]
- Makarova, K.S.; Wolf, Y.I.; Iranzo, J.; Shmakov, S.A.; Alkhnbashi, O.S.; Brouns, S.J.J.; Charpentier, E.; Cheng, D.; Haft, D.H.; Horvath, P.; et al. Evolutionary Classification of CRISPR–Cas Systems: A Burst of Class 2 and Derived Variants. Nat. Rev. Microbiol. 2020, 18, 67–83. [Google Scholar] [CrossRef] [PubMed]
- Colavecchio, A.; D’Souza, Y.; Tompkins, E.; Jeukens, J.; Freschi, L.; Emond-Rheault, J.-G.; Kukavica-Ibrulj, I.; Boyle, B.; Bekal, S.; Tamber, S.; et al. Prophage Integrase Typing Is a Useful Indicator of Genomic Diversity in Salmonella Enterica. Front. Microbiol. 2017, 8, 1283. [Google Scholar] [CrossRef] [PubMed]
- Morris, F.C.; Dexter, C.; Kostoulias, X.; Uddin, M.I.; Peleg, A.Y. The Mechanisms of Disease Caused by Acinetobacter baumannii. Front. Microbiol. 2019, 10, 1601. [Google Scholar] [CrossRef] [PubMed]
- Garnacho-Montero, J.; Timsit, J.-F. Managing Acinetobacter baumannii Infections. Curr. Opin. Infect. Dis. 2019, 32, 69–76. [Google Scholar] [CrossRef]
- Tacconelli, E.; Carrara, E.; Savoldi, A.; Harbarth, S.; Mendelson, M.; Monnet, D.L.; Pulcini, C.; Kahlmeter, G.; Kluytmans, J.; Carmeli, Y.; et al. Discovery, Research, and Development of New Antibiotics: The WHO Priority List of Antibiotic-Resistant Bacteria and Tuberculosis. Lancet Infect. Dis. 2018, 18, 318–327. [Google Scholar] [CrossRef] [PubMed]
- Karah, N.; Samuelsen, Ø.; Zarrilli, R.; Sahl, J.W.; Wai, S.N.; Uhlin, B.E. CRISPR-Cas Subtype I-Fb in Acinetobacter baumannii: Evolution and Utilization for Strain Subtyping. PLoS ONE 2015, 10, e0118205. [Google Scholar] [CrossRef] [PubMed]
- Tyumentseva, M.; Mikhaylova, Y.; Prelovskaya, A.; Tyumentsev, A.; Petrova, L.; Fomina, V.; Zamyatin, M.; Shelenkov, A.; Akimkin, V. Genomic and Phenotypic Analysis of Multidrug-Resistant Acinetobacter baumannii Clinical Isolates Carrying Different Types of CRISPR/Cas Systems. Pathogens 2021, 10, 205. [Google Scholar] [CrossRef]
- Mortensen, K.; Lam, T.J.; Ye, Y. Comparison of CRISPR–Cas Immune Systems in Healthcare-Related Pathogens. Front. Microbiol. 2021, 12, 758782. [Google Scholar] [CrossRef]
- Mangas, E.L.; Rubio, A.; Álvarez-Marín, R.; Labrador-Herrera, G.; Pachón, J.; Pachón-Ibáñez, M.E.; Divina, F.; Pérez-Pulido, A.J. Pangenome of Acinetobacter baumannii Uncovers Two Groups of Genomes, One of Them with Genes Involved in CRISPR/Cas Defence Systems Associated with the Absence of Plasmids and Exclusive Genes for Biofilm Formation. Microb. Genom. 2019, 5. [Google Scholar] [CrossRef]
- Guo, T.; Sun, X.; Li, M.; Wang, Y.; Jiao, H.; Li, G. The Involvement of the Csy1 Gene in the Antimicrobial Resistance of Acinetobacter baumannii. Front. Med. 2022, 9, 797104. [Google Scholar] [CrossRef]
- Touchon, M.; Cury, J.; Yoon, E.-J.; Krizova, L.; Cerqueira, G.C.; Murphy, C.; Feldgarden, M.; Wortman, J.; Clermont, D.; Lambert, T.; et al. The Genomic Diversification of the Whole Acinetobacter Genus: Origins, Mechanisms, and Consequences. Genome Biol. Evol. 2014, 6, 2866–2882. [Google Scholar] [CrossRef] [PubMed]
- Hauck, Y.; Soler, C.; Jault, P.; Mérens, A.; Gérome, P.; Nab, C.M.; Trueba, F.; Bargues, L.; Thien, H.V.; Vergnaud, G.; et al. Diversity of Acinetobacter baumannii in Four French Military Hospitals, as Assessed by Multiple Locus Variable Number of Tandem Repeats Analysis. PLoS ONE 2012, 7, e44597. [Google Scholar] [CrossRef]
- Montaña, S.; Vilacoba, E.; Fernandez, J.S.; Traglia, G.M.; Sucari, A.; Pennini, M.; Iriarte, A.; Centron, D.; Melano, R.G.; Ramírez, M.S. Genomic Analysis of Two Acinetobacter baumannii Strains Belonging to Two Different Sequence Types (ST172 and ST25). J. Glob. Antimicrob. Resist. 2020, 23, 154–161. [Google Scholar] [CrossRef] [PubMed]
- Galani, V.; Papagiannitsis, C.C.; Petinaki, E. First Description of ST409 OXA-23-Producing Acinetobacter baumannii, Carrying a CST8 CRISPR/Cas System, in Central Greece. J. Glob. Antimicrob. Resist. 2020, 22, 137–138. [Google Scholar] [CrossRef]
- Silveira, M.C.; Rocha-de-Souza, C.M.; De Oliveira Santos, I.C.; Pontes, L.D.S.; Oliveira, T.R.T.E.; Tavares-Teixeira, C.B.; Cossatis, N.D.A.; Pereira, N.F.; Da Conceição-Neto, O.C.; Da Costa, B.S.; et al. Genetic Basis of Antimicrobial Resistant Gram-Negative Bacteria Isolated From Bloodstream in Brazil. Front. Med. 2021, 8, 635206. [Google Scholar] [CrossRef]
- Leal, N.C.; Campos, T.L.; Rezende, A.M.; Docena, C.; Mendes-Marques, C.L.; De Sá Cavalcanti, F.L.; Wallau, G.L.; Rocha, I.V.; Cavalcanti, C.L.B.; Veras, D.L.; et al. Comparative Genomics of Acinetobacter baumannii Clinical Strains From Brazil Reveals Polyclonal Dissemination and Selective Exchange of Mobile Genetic Elements Associated With Resistance Genes. Front. Microbiol. 2020, 11, 1176. [Google Scholar] [CrossRef] [PubMed]
- Arndt, D.; Grant, J.R.; Marcu, A.; Sajed, T.; Pon, A.; Liang, Y.; Wishart, D.S. PHASTER: A Better, Faster Version of the PHAST Phage Search Tool. Nucleic Acids Res. 2016, 44, W16–W21. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.; Burmeister, A.R.; Wahl, L.M. Evolution along the Parasitism-Mutualism Continuum Determines the Genetic Repertoire of Prophages. PLoS Comput. Biol. 2020, 16, e1008482. [Google Scholar] [CrossRef]
- Tomida, J.; Morita, Y.; Shibayama, K.; Kikuchi, K.; Sawa, T.; Akaike, T.; Kawamura, Y. Diversity and Microevolution of CRISPR Loci in Helicobacter Cinaedi. PLoS ONE 2017, 12, e0186241. [Google Scholar] [CrossRef] [PubMed]
- Andersen, J.M.; Shoup, M.; Robinson, C.; Britton, R.; Olsen, K.E.P.; Barrangou, R. CRISPR Diversity and Microevolution in Clostridium Difficile. Genome Biol. Evol. 2016, 8, 2841–2855. [Google Scholar] [CrossRef]
- Chia, P.Y.; Sengupta, S.; Kukreja, A.; SL Ponnampalavanar, S.; Ng, O.T.; Marimuthu, K. The Role of Hospital Environment in Transmissions of Multidrug-Resistant Gram-Negative Organisms. Antimicrob. Resist. Infect. Control. 2020, 9, 29. [Google Scholar] [CrossRef]
- Pursey, E.; Dimitriu, T.; Paganelli, F.L.; Westra, E.R.; Van Houte, S. CRISPR-Cas Is Associated with Fewer Antibiotic Resistance Genes in Bacterial Pathogens. Philos. Trans. R. Soc. B. 2022, 377, 20200464. [Google Scholar] [CrossRef]
- Horvath, P.; Coûté-Monvoisin, A.-C.; Romero, D.A.; Boyaval, P.; Fremaux, C.; Barrangou, R. Comparative Analysis of CRISPR Loci in Lactic Acid Bacteria Genomes. Int. J. Food Microbiol. 2009, 131, 62–70. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Ye, Y. Not All Predicted CRISPR–Cas Systems Are Equal: Isolated Cas Genes and Classes of CRISPR like Elements. BMC Bioinform. 2017, 18, 92. [Google Scholar] [CrossRef] [PubMed]
- Nobrega, F.L.; Walinga, H.; Dutilh, B.E.; Brouns, S.J.J. Prophages Are Associated with Extensive CRISPR–Cas Auto-Immunity. Nucleic Acids Res. 2020, 48, 12074–12084. [Google Scholar] [CrossRef] [PubMed]
- Luz, A.C.D.O.; Da Silva, J.M.A.; Rezende, A.M.; De Barros, M.P.S.; Leal-Balbino, T.C. Analysis of Direct Repeats and Spacers of CRISPR/Cas Systems Type I-F in Brazilian Clinical Strains of Pseudomonas Aeruginosa. Mol. Genet. Genom. 2019, 294, 1095–1105. [Google Scholar] [CrossRef] [PubMed]
- Barros, M.P.S.; França, C.T.; Lins, R.H.F.B.; Santos, M.D.V.; Silva, E.J.; Oliveira, M.B.M.; Silveira-Filho, V.M.; Rezende, A.M.; Balbino, V.Q.; Leal-Balbino, T.C. Dynamics of CRISPR Loci in Microevolutionary Process of Yersinia Pestis Strains. PLoS ONE 2014, 9, e108353. [Google Scholar] [CrossRef]
- Godde, J.S.; Bickerton, A. The Repetitive DNA Elements Called CRISPRs and Their Associated Genes: Evidence of Horizontal Transfer Among Prokaryotes. J. Mol. Evol. 2006, 62, 718–729. [Google Scholar] [CrossRef]
- Edgar, R.; Qimron, U. The Escherichia Coli CRISPR System Protects from λ Lysogenization, Lysogens, and Prophage Induction. J. Bacteriol. 2010, 192, 6291–6294. [Google Scholar] [CrossRef] [PubMed]
- McGinn, J.; Marraffini, L.A. CRISPR-Cas Systems Optimize Their Immune Response by Specifying the Site of Spacer Integration. Mol. Cell. 2016, 64, 616–623. [Google Scholar] [CrossRef] [PubMed]
- Ou, Y.; McInerney, J.O. High Frequency of Dynamic Rearrangements in Crispr Loci. Evol. Biol. 2022. [Google Scholar] [CrossRef]
- Pillonetto, M.; Arend, L.; Vespero, E.C.; Pelisson, M.; Chagas, T.P.G.; Carvalho-Assef, A.P.D.; Asensi, M.D. First Report of NDM-1-Producing Acinetobacter baumannii Sequence Type 25 in Brazil. Antimicrob. Agents Chemother. 2014, 58, 7592–7594. [Google Scholar] [CrossRef] [PubMed]
- Camargo, C.H.; Cunha, M.P.V.; De Barcellos, T.A.F.; Bueno, M.S.; Bertani, A.M.D.J.; Dos Santos, C.A.; Nagamori, F.O.; Takagi, E.H.; Chimara, E.; De Carvalho, E.; et al. Genomic and Phenotypic Characterisation of Antimicrobial Resistance in Carbapenem-Resistant Acinetobacter baumannii Hyperendemic Clones CC1, CC15, CC79 and CC25. Int. J. Antimicrob. Agents 2020, 56, 106195. [Google Scholar] [CrossRef]
- Sahl, J.W.; Del Franco, M.; Pournaras, S.; Colman, R.E.; Karah, N.; Dijkshoorn, L.; Zarrilli, R. Phylogenetic and Genomic Diversity in Isolates from the Globally Distributed Acinetobacter baumannii ST25 Lineage. Sci. Rep. 2015, 5, 15188. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Li, Y.; Gorgé, O.; Platonov, M.E.; Yan, Y.; Guo, Z.; Pourcel, C.; Dentovskaya, S.V.; Balakhonov, S.V.; Wang, X.; et al. Insight into Microevolution of Yersinia Pestis by Clustered Regularly Interspaced Short Palindromic Repeats. PLoS ONE 2008, 3, e2652. [Google Scholar] [CrossRef]
- Shariat, N.; Timme, R.E.; Pettengill, J.B.; Barrangou, R.; Dudley, E.G. Characterization and Evolution of Salmonella CRISPR-Cas Systems. Microbiology 2015, 161, 374–386. [Google Scholar] [CrossRef]
- Rafei, R.; Osman, M.; Dabboussi, F.; Hamze, M. Update on the Epidemiological Typing Methods for Acinetobacter baumannii. Future Microbiol. 2019, 14, 1065–1080. [Google Scholar] [CrossRef] [PubMed]
- Grissa, I.; Vergnaud, G.; Pourcel, C. CRISPRFinder: A Web Tool to Identify Clustered Regularly Interspaced Short Palindromic Repeats. Nucleic Acids Res. 2007, 35, W52–W57. [Google Scholar] [CrossRef] [PubMed]
- Semenova, E.; Nagornykh, M.; Pyatnitskiy, M.; Artamonova, I.I.; Severinov, K. Analysis of CRISPR System Function in Plant Pathogen Xanthomonas Oryzae. FEMS Microbiol. Lett. 2009, 296, 110–116. [Google Scholar] [CrossRef]
- Berriman, M. Viewing and Annotating Sequence Data with Artemis. Brief. Bioinform. 2003, 4, 124–132. [Google Scholar] [CrossRef]
- Wang, J.; Dai, W.; Li, J.; Li, Q.; Xie, R.; Zhang, Y.; Stubenrauch, C.; Lithgow, T. AcrHub: An Integrative Hub for Investigating, Predicting and Mapping Anti-CRISPR Proteins. Nucleic Acids Res. 2021, 49, D630–D638. [Google Scholar] [CrossRef]
- Yi, H.; Huang, L.; Yang, B.; Gomez, J.; Zhang, H.; Yin, Y. AcrFinder: Genome Mining Anti-CRISPR Operons in Prokaryotes and Their Viruses. Nucleic Acids Res. 2020, 48, W358–W365. [Google Scholar] [CrossRef]
- Biswas, A.; Gagnon, J.N.; Brouns, S.J.J.; Fineran, P.C.; Brown, C.M. CRISPRTarget: Bioinformatic Prediction and Analysis of CrRNA Targets. RNA Biol. 2013, 10, 817–827. [Google Scholar] [CrossRef]
- Gaiarsa, S.; Batisti Biffignandi, G.; Esposito, E.P.; Castelli, M.; Jolley, K.A.; Brisse, S.; Sassera, D.; Zarrilli, R. Comparative Analysis of the Two Acinetobacter baumannii Multilocus Sequence Typing (MLST) Schemes. Front. Microbiol. 2019, 10, 930. [Google Scholar] [CrossRef] [PubMed]
- Couvin, D.; Bernheim, A.; Toffano-Nioche, C.; Touchon, M.; Michalik, J.; Néron, B.; Rocha, E.P.C.; Vergnaud, G.; Gautheret, D.; Pourcel, C. CRISPRCasFinder, an Update of CRISRFinder, Includes a Portable Version, Enhanced Performance and Integrates Search for Cas Proteins. Nucleic Acids Res. 2018, 46, W246–W251. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular Evolutionary Genetics Analysis Version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef]
- Dereeper, A.; Guignon, V.; Blanc, G.; Audic, S.; Buffet, S.; Chevenet, F.; Dufayard, J.-F.; Guindon, S.; Lefort, V.; Lescot, M.; et al. Phylogeny.Fr: Robust Phylogenetic Analysis for the Non-Specialist. Nucleic Acids Res. 2008, 36, W465–W469. [Google Scholar] [CrossRef]
- Treangen, T.J.; Ondov, B.D.; Koren, S.; Phillippy, A.M. The Harvest Suite for Rapid Core-Genome Alignment and Visualization of Thousands of Intraspecific Microbial Genomes. Genome Biol. 2014, 15, 524. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Bork, P. Interactive Tree Of Life (ITOL): An Online Tool for Phylogenetic Tree Display and Annotation. Bioinformatics 2007, 23, 127–128. [Google Scholar] [CrossRef] [PubMed]
- Seemann, T. Prokka: Rapid Prokaryotic Genome Annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Isolates | Size in Kb | Prophage |
|---|---|---|
| Acb_1 | 21.1 | PHAGE_Haemop_SuMu_NC_019455 |
| Acb_4 | 17.9 | PHAGE_Acinet_Bphi_B1251_NC_019541 |
| 39.4 | PHAGE_Haemop_SuMu_NC_019455 | |
| 34.6 | PHAGE_Haemop_SuMu_NC_019455 | |
| Acb_5 | 43.4 | PHAGE_Acinet_Bphi_B1251_NC_019541 |
| 31.3 | PHAGE_Pseudo_B3_NC_006548 | |
| 51.4 | PHAGE_Acinet_vB_AbaS_TRS1_NC_031098 | |
| 38.2 | PHAGE_Acinet_vB_AbaS_TRS1_NC_031098 | |
| Acb_8 | 21.1 | PHAGE_Mannhe_vB_MhM_3927AP2_NC_028766 |
| Acb_21 | 39.3 | PHAGE_Mannhe_vB_MhM_3927AP2_NC_028766 |
| 37.4 | PHAGE_Pelagi_HTVC010P_NC_020481 | |
| 42.6 | PHAGE_Acinet_Bphi_B1251_NC_019541 | |
| Acb_24 | 38 | PHAGE_Acinet_vB_AbaS_TRS1_NC_031098 |
| 21.1 | PHAGE_Haemop_SuMu_NC_019455 | |
| 42.8 | PHAGE_Acinet_Bphi_B1251_NC_019541 | |
| 66.9 | PHAGE_Psychr_Psymv2_NC_023734 | |
| Acb_29 | 17.9 | PHAGE_Haemop_SuMu_NC_019455 |
| 43.6 | PHAGE_Acinet_vB_AbaS_TRS1_NC_031098 | |
| 10.5 | PHAGE_Psychr_Psymv2_NC_023734 | |
| 23.4 | PHAGE_Mannhe_vB_MhM_3927AP2_NC_028766 | |
| Acb_33 | 18.1 | PHAGE_Psychr_Psymv2_NC_023734 |
| 28.9 | PHAGE_Mannhe_vB_MhM_3927AP2_NC_028766 | |
| Acb_35 | 16.6 | PHAGE_Psychr_Psymv2_NC_023734 |
| 39.8 | PHAGE_Mannhe_vB_MhM_3927AP2_NC_028766 | |
| Acb_36 | 21.9 | PHAGE_Mannhe_vB_MhM_3927AP2_NC_028766 |
| Acb_38 | 24.3 | PHAGE_Haemop_SuMu_NC_019455 |
| 16.6 | PHAGE_Psychr_Psymv2_NC_023734 | |
| Acb_41 | - | - |
| Acb_44 | 36 | PHAGE_Psychr_Psymv2_NC_023734 |
| 38.5 | PHAGE_Pseudo_Dobby_NC_048109 | |
| 36.9 | PHAGE_Haemop_SuMu_NC_019455 | |
| Acb_47 | 38.2 | PHAGE_Escher_ECP1_NC_049926 |
| 13 | PHAGE_Pseudo_phiCTX_NC_003278 | |
| 41.6 | PHAGE_Mannhe_vB_MhM_3927AP2_NC_028766 | |
| 7 | PHAGE_Mannhe_vB_MhM_3927AP2_NC_028766 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Silva, A.M.A.; Luz, A.C.O.; Xavier, K.V.M.; Barros, M.P.S.; Alves, H.B.; Batista, M.V.A.; Leal-Balbino, T.C. Analysis of CRISPR/Cas Genetic Structure, Spacer Content and Molecular Epidemiology in Brazilian Acinetobacter baumannii Clinical Isolates. Pathogens 2023, 12, 764. https://doi.org/10.3390/pathogens12060764
Silva AMA, Luz ACO, Xavier KVM, Barros MPS, Alves HB, Batista MVA, Leal-Balbino TC. Analysis of CRISPR/Cas Genetic Structure, Spacer Content and Molecular Epidemiology in Brazilian Acinetobacter baumannii Clinical Isolates. Pathogens. 2023; 12(6):764. https://doi.org/10.3390/pathogens12060764
Chicago/Turabian StyleSilva, Adrianne M. A., Ana C. O. Luz, Keyla V. M. Xavier, Maria P. S. Barros, Hirisleide B. Alves, Marcus V. A. Batista, and Tereza C. Leal-Balbino. 2023. "Analysis of CRISPR/Cas Genetic Structure, Spacer Content and Molecular Epidemiology in Brazilian Acinetobacter baumannii Clinical Isolates" Pathogens 12, no. 6: 764. https://doi.org/10.3390/pathogens12060764