
The Microbial Genetic Diversity and Succession Associated with Processing Waters at Different Broiler Processing Stages in an Abattoir in Australia
 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. Sample Preparation
2.2.1. Culture-Based Technique
2.2.2. 16S rRNA Amplicon Sequencing
3. Results and Discussion
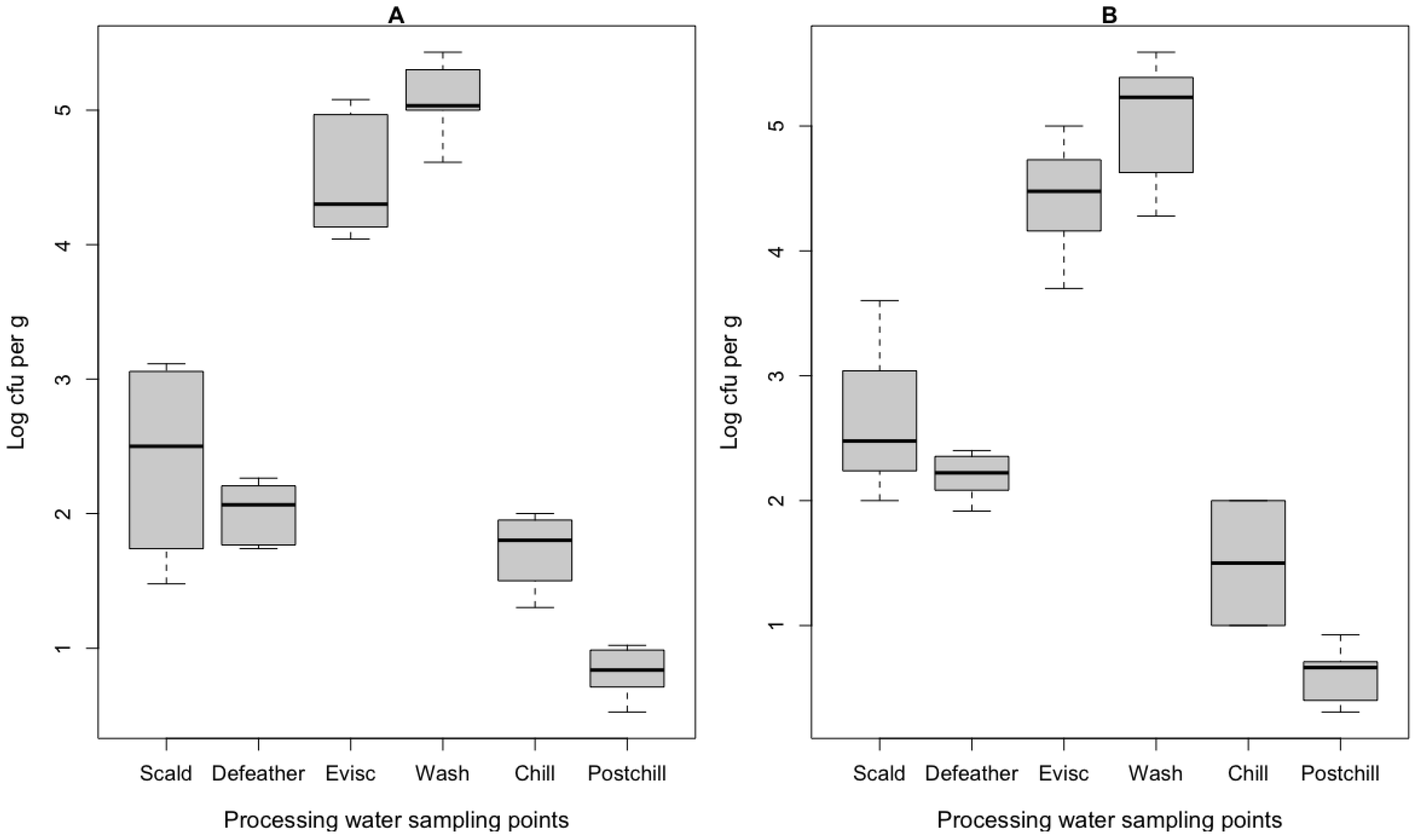
3.1. Culture-Dependent Bacteria Assessment
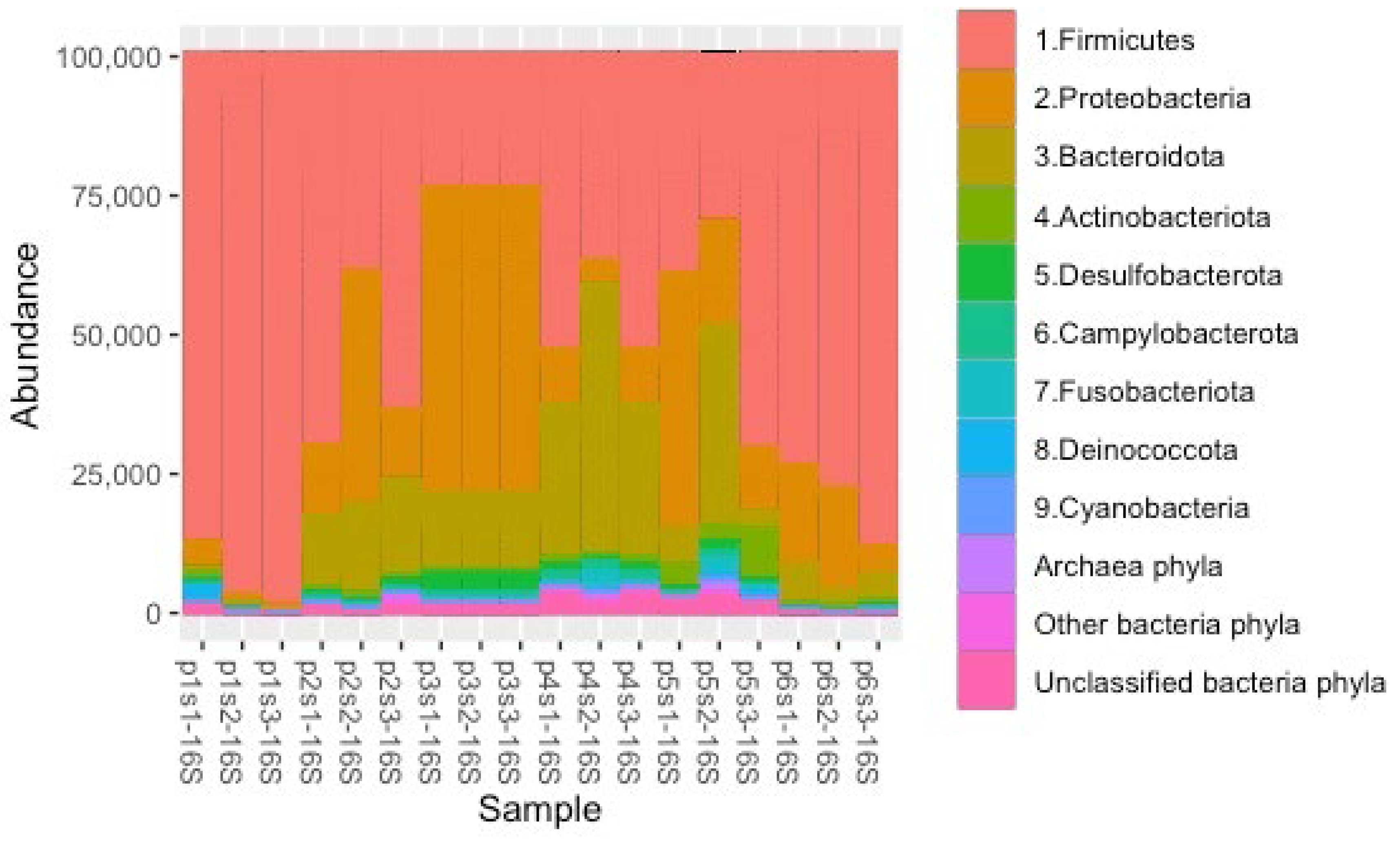
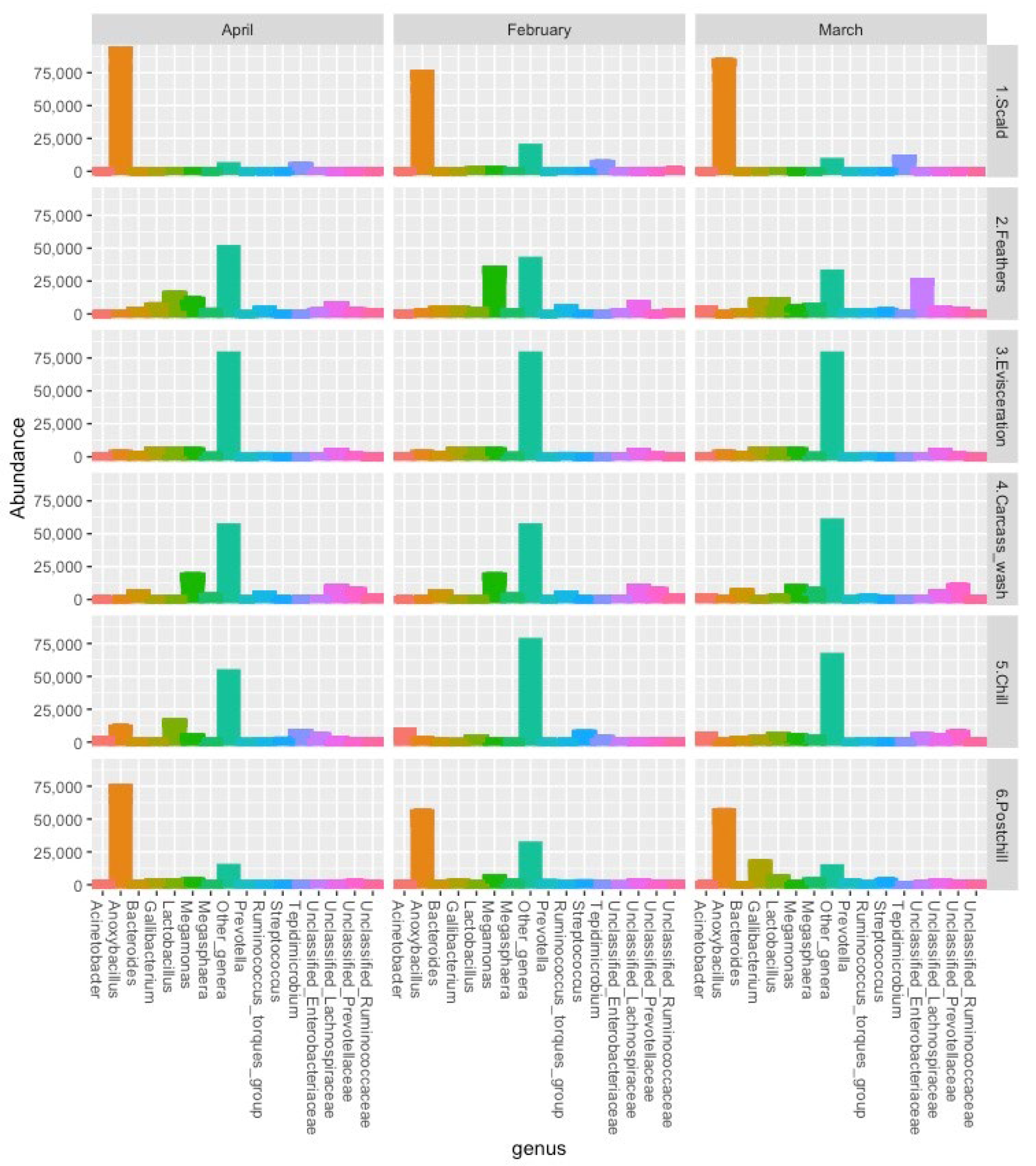
3.2. ASV Abundance
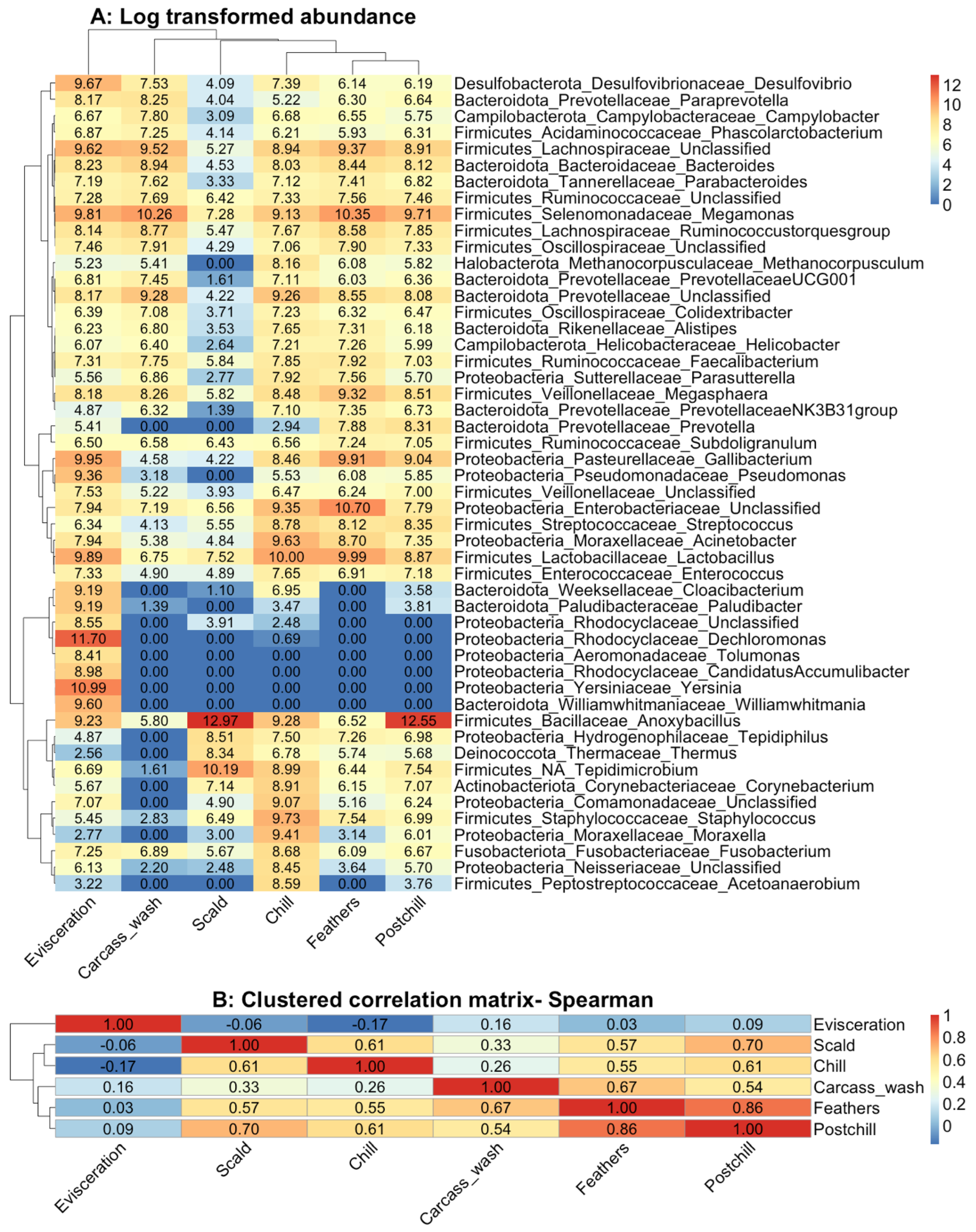
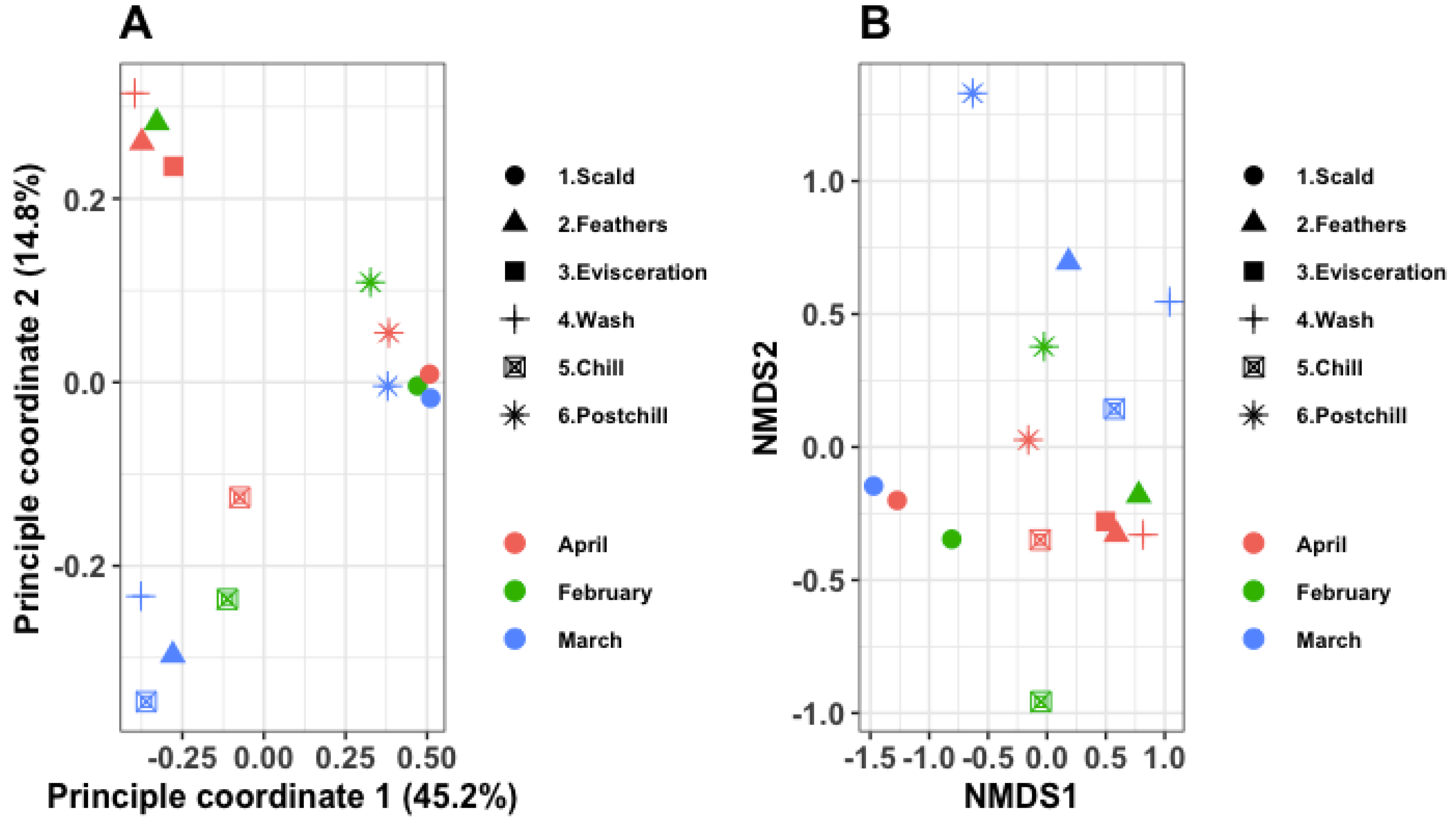
3.3. Cluster Analysis
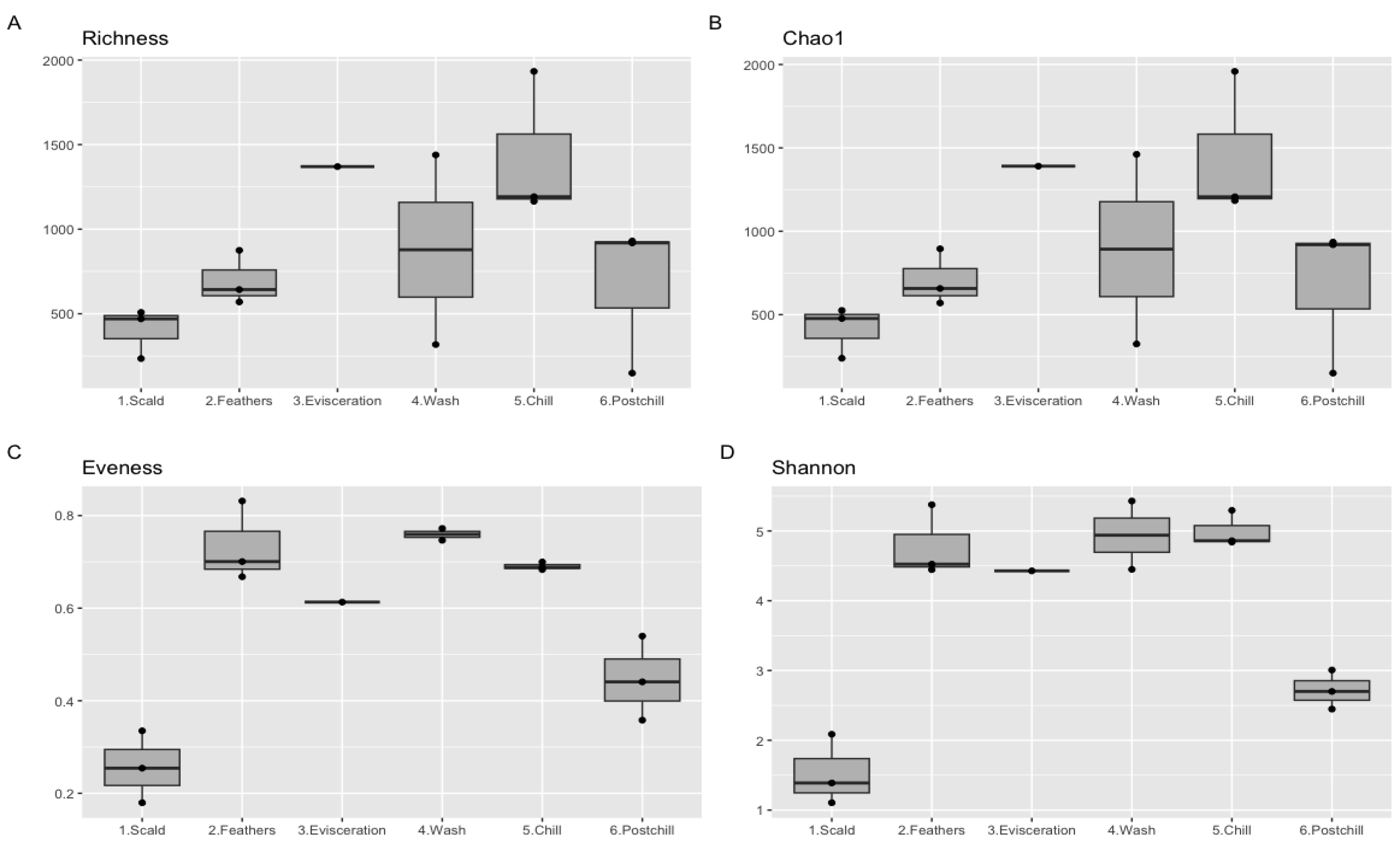
3.4. Alpha Diversity
3.5. Beta Diversity
4. Conclusions and Recommendations
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- OECD. Meat Consumption (Indicator). Available online: https://www.oecd-ilibrary.org/agriculture-and-food/meat-consumption/indicator/english_fa290fd0-en (accessed on 18 November 2021).
- Marmion, M.; Ferone, M.T.; Whyte, P.; Scannell, A.G.M.M. The Changing Microbiome of Poultry Meat; from Farm to Fridge. Food Microbiol. 2021, 99, 103823. [Google Scholar] [CrossRef] [PubMed]
- Rajapaksha, P.; Elbourne, A.; Gangadoo, S.; Brown, R.; Cozzolino, D.; Chapman, J. A Review of Methods for the Detection of Pathogenic Microorganisms. Analyst 2019, 144, 396–411. [Google Scholar] [CrossRef] [PubMed]
- Váradi, L.; Luo, J.L.; Hibbs, D.E.; Perry, J.D.; Anderson, R.J.; Orenga, S.; Groundwater, P.W. Methods for the Detection and Identification of Pathogenic Bacteria: Past, Present, and Future. Chem. Soc. Rev. 2017, 46, 4818–4832. [Google Scholar] [CrossRef] [PubMed]
- Berrang, M.E.; Gamble, G.R.; Hinton, A.; Johnston, J.J. Neutralization of Residual Antimicrobial Processing Chemicals in Broiler Carcass Rinse for Improved Detection of Campylobacter. J. Appl. Poult. Res. 2018, 27, 299–303. [Google Scholar] [CrossRef]
- Chen, G.; Chen, C.; Lei, Z. Meta-Omics Insights in the Microbial Community Profiling and Functional Characterization of Fermented Foods. Trends Food Sci. Technol. 2017, 65, 23–31. [Google Scholar] [CrossRef]
- Overney, A.; Jacques-André-Coquin, J.; Ng, P.; Carpentier, B.; Guillier, L.; Firmesse, O. Impact of Environmental Factors on the Culturability and Viability of Listeria Monocytogenes under Conditions Encountered in Food Processing Plants. Int. J. Food Microbiol. 2017, 244, 74–81. [Google Scholar] [CrossRef]
- Ronholm, J.; Nasheri, N.; Petronella, N.; Pagotto, F. Navigating Microbiological Food Safety in the Era of Whole-Genome Sequencing. Clin. Microbiol. Rev. 2016, 29, 837–857. [Google Scholar] [CrossRef] [Green Version]
- Feye, K.M.; Thompson, D.R.; Rothrock, M.J.; Kogut, M.H.; Ricke, S.C. Poultry Processing and the Application of Microbiome Mapping. Poult. Sci. 2020, 99, 678–688. [Google Scholar] [CrossRef]
- Handley, J.A.; Park, S.H.; Kim, S.A.; Ricke, S.C. Microbiome Profiles of Commercial Broilers through Evisceration and Immersion Chilling during Poultry Slaughter and the Identification of Potential Indicator Microorganisms. Front. Microbiol. 2018, 9, 345. [Google Scholar] [CrossRef]
- Cao, Y.; Fanning, S.; Proos, S.; Jordan, K.; Srikumar, S. A Review on the Applications of next Generation Sequencing Technologies as Applied to Food-Related Microbiome Studies. Front. Microbiol. 2017, 8, 1829. [Google Scholar] [CrossRef] [Green Version]
- Xu, J. Microbial Ecology in the Age of Genomics and Metagenomics: Concepts, Tools, and Recent Advances. Mol. Ecol. 2006, 15, 1713–1731. [Google Scholar] [CrossRef] [PubMed]
- Franco-Duarte, R.; Černáková, L.; Kadam, S.; Kaushik, K.S.; Salehi, B.; Bevilacqua, A.; Corbo, M.R.; Antolak, H.; Dybka-Stępień, K.; Leszczewicz, M.; et al. Advances in Chemical and Biological Methods to Identify Microorganisms—From Past to Present. Microorganisms 2019, 7, 130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Amore, R.; Ijaz, U.Z.; Schirmer, M.; Kenny, J.G.; Gregory, R.; Darby, A.C.; Shakya, M.; Podar, M.; Quince, C.; Hall, N. A Comprehensive Benchmarking Study of Protocols and Sequencing Platforms for 16S RRNA Community Profiling. BMC Genom. 2016, 17, 55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rouger, A.; Tresse, O.; Zagorec, M. Bacterial Contaminants of Poultry Meat: Sources, Species, and Dynamics. Microorganisms 2017, 5, 50. [Google Scholar] [CrossRef] [Green Version]
- Ricke, S.C.; Dittoe, D.K.; Brown, J.A.; Thompson, D.R. Practical Opportunities for Microbiome Analyses and Bioinformatics in Poultry Processing. Poult. Sci. 2022, 101, 101787. [Google Scholar] [CrossRef]
- Van Reckem, E.; De Vuyst, L.; Weckx, S.; Leroy, F. Next-Generation Sequencing to Enhance the Taxonomic Resolution of the Microbiological Analysis of Meat and Meat-Derived Products. Curr. Opin. Food Sci. 2021, 37, 58–65. [Google Scholar] [CrossRef]
- Kim, S.A.; Park, S.H.; Lee, S.L.; Owens, C.M.; Ricke, S.C.; Ae Kim, S.; Hong Park, S.; In Lee, S.; Owens, C.M.; Ricke, S.C. Assessment of Chicken Carcass Microbiome Responses during Processing in the Presence of Commercial Antimicrobials Using a Next Generation Sequencing Approach. Sci. Rep. 2017, 7, 43354. [Google Scholar] [CrossRef] [Green Version]
- Rothrock, M.J.; Locatelli, A.; Glenn, T.C.; Thomas, J.C.; Caudill, A.C.; Kiepper, B.H.; Hiett, K.L. Assessing the Microbiomes of Scalder and Chiller Tank Waters throughout a Typical Commercial Poultry Processing Day. Poult. Sci. 2016, 95, 2372–2382. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.H.; Fegan, N.; Kocharunchitt, C.; Bowman, J.P.; Duffy, L.L. Changes of the Bacterial Community Diversity on Chicken Carcasses through an Australian Poultry Processing Line. Food Microbiol. 2020, 86, 103350. [Google Scholar] [CrossRef]
- Jabari, L.; Gannoun, H.; Khelifi, E.; Cayol, J.L.; Godon, J.J.; Hamdi, M.; Fardeaub, M.L. Bacterial Ecology of Abattoir Wastewater Treated by an Anaerobic Digestor. Braz. J. Microbiol. 2016, 47, 73–84. [Google Scholar] [CrossRef]
- Rothrock, M.J.; Hiett, K.L.; Kiepper, B.H.; Ingram, K.; Hinton, A. Quantification of Zoonotic Bacterial Pathogens within Commercial Poultry Processing Water Samples Using Droplet Digital PCR. Adv. Microbiol. 2013, 3, 403–411. [Google Scholar] [CrossRef] [Green Version]
- Palatsi, J.; Viñas, M.; Guivernau, M.; Fernandez, B.; Flotats, X. Anaerobic Digestion of Slaughterhouse Waste: Main Process Limitations and Microbial Community Interactions. Bioresour. Technol. 2011, 102, 2219–2227. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.E.; Lee, J.J.; Lee, M.J.; Kim, B.S. Analysis of Microbiome in Raw Chicken Meat from Butcher Shops and Packaged Products in South Korea to Detect the Potential Risk of Foodborne Illness. Food Res. Int. 2019, 122, 517–527. [Google Scholar] [CrossRef] [PubMed]
- Morgan, M.; Anders, S.; Lawrence, M.; Aboyoun, P.; Pagès, H.; Gentleman, R. ShortRead: A Bioconductor Package for Input, Quality Assessment and Exploration of High-Throughput Sequence Data. Bioinformatics 2009, 25, 2607–2608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-Resolution Sample Inference from Illumina Amplicon Data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Peng, Z.; Li, P.; Mao, Y.; Shen, R.; Tao, R.; Diao, X.; Liu, L.; Zhao, Y.; Luo, X. Complex Internal Microstructure of Feather Follicles on Chicken Skin Promotes the Bacterial Cross-Contamination of Carcasses during the Slaughtering Process. Front. Microbiol. 2020, 11, 571913. [Google Scholar] [CrossRef]
- Wang, H.; Qin, X.; Mi, S.; Li, X.; Wang, X.; Yan, W.; Zhang, C. Contamination of Yellow-Feathered Broiler Carcasses: Microbial Diversity and Succession during Processing. Food Microbiol. 2019, 83, 18–26. [Google Scholar] [CrossRef]
- Song, X.; Wang, H.; Xu, X. Investigation of Microbial Contamination in a Chicken Slaughterhouse Environment. J. Food Sci. 2021, 86, 3598–3610. [Google Scholar] [CrossRef]
- Chen, S.H.; Fegan, N.; Kocharunchitt, C.; Bowman, J.P.; Duffy, L.L. Impact of Poultry Processing Operating Parameters on Bacterial Transmission and Persistence on Chicken Carcasses and Their Shelf Life. Appl. Environ. Microbiol. 2020, 86, e00594-20. [Google Scholar] [CrossRef]
- Kunert-Filho, H.C.; Furian, T.Q.; Sesterhenn, R.; Chitolina, G.Z.; Willsmann, D.E.; Borges, K.A.; Salle, C.T.P.; de Moraes, H.L.S.; do Nascimento, V.P. Bacterial Community Identification in Poultry Carcasses Using High-Throughput next Generation Sequencing. Int. J. Food Microbiol. 2022, 364, 109533. [Google Scholar] [CrossRef]
- Abellan-Schneyder, I.; Matchado, M.S.; Reitmeier, S.; Sommer, A.; Sewald, Z.; Baumbach, J.; List, M.; Neuhaus, K. Primer, Pipelines, Parameters: Issues in 16S RRNA Gene Sequencing. mSphere 2021, 6, e01202-20. [Google Scholar] [CrossRef] [PubMed]
- Hauge, S.J.; Johannessen, G.S.; Haverkamp, T.H.A.; Bjørkøy, S.; Llarena, A.K.; Spilsberg, B.; Leithaug, M.; Økland, M.; Holthe, J.; Røtterud, O.J.; et al. Assessment of Poultry Process Hygiene and Bacterial Dynamics along Two Broiler Slaughter Lines in Norway. Food Control 2023, 146, 109526. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genus | Scald | Feathers | Evisceration | Carcass wash | Chill | Post-Chill |
|---|---|---|---|---|---|---|
| Anoxybacillus | 429,789 | 678 | 10,239 | 328 | 10,714 | 283,148 |
| Megamonas | 1444 | 31,374 | 18,165 | 28,700 | 9187 | 16,475 |
| Gallibacterium | 67 | 20,199 | 20,904 | 97 | 4705 | 8444 |
| Unclassified Lachnospiraceae | 193 | 11,735 | 15,081 | 13,627 | 7655 | 7382 |
| Lactobacillus | 1842 | 21,819 | 19,797 | 855 | 22,119 | 7101 |
| Megasphaera | 337 | 11,142 | 3585 | 3873 | 4838 | 4948 |
| Streptococcus | 256 | 3358 | 564 | 61 | 6479 | 4211 |
| Prevotella | 0 | 2648 | 222 | 0 | 18 | 4074 |
| Bacteroides | 92 | 4622 | 3738 | 7598 | 3068 | 3352 |
| Unclassified Prevotellaceae | 67 | 5180 | 3531 | 10,735 | 10,526 | 3216 |
| Ruminococcus (torques group) | 236 | 5327 | 3435 | 6453 | 2152 | 2553 |
| Unclassified Enterobacteriaceae | 704 | 44,191 | 2796 | 1325 | 11,554 | 2425 |
| Tepidimicrobium | 26,575 | 627 | 801 | 4 | 7988 | 1886 |
| Unclassified Ruminococcaceae | 611 | 1916 | 1455 | 2191 | 1528 | 1737 |
| Acinetobacter | 125 | 6000 | 2811 | 216 | 15,285 | 1552 |
| Unclassified Oscillospiraceae | 72 | 2702 | 1728 | 2717 | 1165 | 1522 |
| Enterococcus | 132 | 1005 | 1527 | 133 | 2094 | 1310 |
| Catenibacterium | 0 | 1249 | 105 | 0 | 9 | 1216 |
| Unclassified Moraxellaceae | 61 | 739 | 102 | 36 | 1729 | 1184 |
| Corynebacterium | 1256 | 468 | 288 | 0 | 7427 | 1181 |
| Subdoligranulum | 617 | 1387 | 666 | 722 | 704 | 1152 |
| Faecalibacterium | 344 | 2757 | 1488 | 2325 | 2572 | 1126 |
| Unclassified Veillonellaceae | 50 | 510 | 1866 | 184 | 642 | 1097 |
| Staphylococcus | 659 | 1890 | 231 | 16 | 16,877 | 1090 |
| Tepidiphilus | 4960 | 1418 | 129 | 0 | 1806 | 1078 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gichure, J.N.; Coorey, R.; Njage, P.M.K.; Dykes, G.A.; Muema, E.K.; Buys, E.M. The Microbial Genetic Diversity and Succession Associated with Processing Waters at Different Broiler Processing Stages in an Abattoir in Australia. Pathogens 2023, 12, 488. https://doi.org/10.3390/pathogens12030488
Gichure JN, Coorey R, Njage PMK, Dykes GA, Muema EK, Buys EM. The Microbial Genetic Diversity and Succession Associated with Processing Waters at Different Broiler Processing Stages in an Abattoir in Australia. Pathogens. 2023; 12(3):488. https://doi.org/10.3390/pathogens12030488
Chicago/Turabian StyleGichure, Josphat Njenga, Ranil Coorey, Patrick Murigu Kamau Njage, Gary A. Dykes, Esther K. Muema, and Elna M. Buys. 2023. "The Microbial Genetic Diversity and Succession Associated with Processing Waters at Different Broiler Processing Stages in an Abattoir in Australia" Pathogens 12, no. 3: 488. https://doi.org/10.3390/pathogens12030488